Key Words: Monitoring Aseptik Isolator RABS risikobasiert
Risikobasierte Festlegung von Monitoringpunkten im aseptischen Herstellbereich
Korrespondenz:
Christian Gavranovic, PPT Pharma Process Technology GmbH, Neue Mainzer Str. 66–68, 60311 Frankfurt am Main; E-Mail: christian.gavranovic@pp-technology.de
 | Christian Gavranovic Christian Gavranovic ist studierter Maschinenbau-Ingenieur. Er startete seine berufliche Laufbahn bei der Boehringer Ingelheim GmbH als Process Engineer. Nach seinem Wechsel zur Biotest AG betreute er zuerst den Bereich der aseptischen Abfüllung und Gefriertrocknung als Quality Assurance Manager. Anschließend unterstützte er die Abteilung der Endkonfektionierung als Betriebsassistent. Ab 2019 begleitete er als Discipline-Lead Fill Finish/Aseptic Technologies Unternehmen bei der Planung, Auslegung und Realisierung ihrer aseptischen Prozesse. Berufsbegleitend konnte er sein Studium zum Master in Quality Management abschließen und ist seit Apr. 2021 als Leiter der Abteilung Quality and Compliance bei der PPT Pharma Process Technology GmbH tätig. |
 | Gerald Mathe Gerald Mathe verfügt über mehr als 30 Jahre Erfahrung in der Pharmaindustrie. Schwerpunkte waren dabei die Instandhaltung, Produktion – v. a. der Sterilherstellung, Entwicklung von technischen Sonderprozessen und der Planung und Umsetzung von Investitionsprojekten. Von 1997–2017 war Mathe bei Boehringer Ingelheim in verschiedenen Funktionen beschäftigt. Seit 2017 ist er im Bereich der Engineering Dienstleistungen tätig. Heute ist er bei der PPT Pharma Process Technology GmbH in Frankfurt als CEO und Aseptic Expert für Fragestellungen rund um die Sterilherstellung vom Projekt bis zum laufenden Betrieb tätig. |
Zusammenfassung
Die Sterilität ist das wichtigste Qualitätsmerkmal der Umgebung der Reinraumklasse A/ISO 5 zur Fertigung aseptischer Produkte. Die Hersteller von aseptischen Darreichungsformen müssen diese Qualität gewährleisten und die hierfür „kritischen Einflussfaktoren“ überwachen. Die Bewertung und Festlegung der Monitoringpunkte muss hierbei schon in einer frühen Projektphase erfolgen. Bei der risikobasierten Festlegung der Monitoringpunkte müssen unterschiedliche regulatorische Anforderungen berücksichtigt werden, die im Rahmen der Design-Phase des RABS- oder Isolator-Systems über die Design-FMEA und Mock-up bestätigt werden.
1. Einleitung
Die Überwachung der Sterilität der Produktionsumgebung der Reinraumklasse A/ISO 5 erfolgt über ein aktives und passives Monitoringsystem der kritischen Einflussfaktoren. Bei diesen handelt es sich um Luft- und Oberflächenkeime, Partikel, Luftgeschwindigkeit, Druck, Feuchte und Temperatur. Da es sich bei den Räumen der Reinraumklasse A/ISO 5 um Restricted-Area-Barrier-Systems (RABS) oder Isolator-Technologien handelt, sind die Monitoringpunkte fester Bestandteil der Anlage. Aus diesem Grund müssen Bewertung und Festlegung der Monitoringpunkte schon in einer frühen Projektphase erfolgen und im Rahmen des Design-Review bestätigt werden.
In der hier vorgestellten Fallstudie wird die risikobasierte Festlegung der aktiven Monitoringpunkte für Luftkeime und Partikel beschrieben. Bei der betrachteten Sterilabfüllung handelt es sich um eine Abfüllanlage mit RABS-Aufbau und nachgeschalteter Gefriertrocknung, die über manuelle Transportwagen beladen wird. Hierfür werden zuerst die regulatorischen Anforderungen aus dem Good-Manufacturing-Practice(GMP)-Leitfaden der EU, der Guidance der U.S. Food and Drug Administration (FDA) und der ISO-Norm durchleuchtet, die Kernaussagen zusammengefasst und in Form von User-Requirement-Specification(URS)-Punkten umgesetzt. Nach der Beschreibung des Produktflusses und der Prozesse in der aseptischen Kernzone wird die risikobasierte Festlegung der Monitoringpunkte beschrieben und wie diese im Rahmen der Design-Phase über die Failure Mode and Effects Analysis (FMEA) des Designs und Mock-up bestätigt werden.
2. Regulatorische Grundlagen/Normen
Bei der Herstellung von sterilen Produkten müssen bestimmte Anforderungen erfüllt werden, um das Risiko einer Kontamination des Produkts mit Partikeln oder Mikroorganismen zu minimieren. Um die Unversehrtheit des sterilen Produkts zu gewährleisten, müssen Überwachungssysteme installiert werden, die Partikel oder luftgetragene Mikroorganismen erkennen und quantifizieren können. Die Positionierung dieser Überwachungssysteme muss durch eine Risikobewertung bestimmt werden.
Hierzu bieten die folgenden Guidelines und Normen eine Anleitung für die richtige Positionierung der Überwachungssysteme im Bereich der Reinraumklasse (RRK) A:
Kapitel 2.1: GMP Annex 1 – Herstellung steriler Arzneimittel
Kapitel 2.2: PIC/S PI 007-6 – Recommendation on the Validation of Aseptic Processes
Kapitel 2.3: EN ISO 14644-1:2015: Reinräume und zugehörige Reinraumbereiche – Teil 1: Klassifizierung der Luftreinheit anhand der Partikelkonzentration
Kapitel 2.4: EN ISO 14644-2:2015: Reinräume und zugehörige Reinraumbereiche – Teil 2: Überwachung zum Nachweis der Reinraumleistung bzgl. Luftreinheit anhand der Partikelkonzentration
Kapitel 2.5: VDI-Richtlinien Blatt 3.1: Reinraumtechnik – Messtechnik in der Reinraumluft – Monitoring
Kapitel 2.6: EN ISO 14698-1:2003: Reinräume und zugehörige Reinraumbereiche. Biokontaminationskontrolle – Teil 1: Allgemeine Grundlagen
Kapitel 2.7: ZLG Aide-mémoire 07120606: Überwachung von Sterilherstellern
Kapitel 2.8: FDA Guidance for Industry Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice
2.1 EU-GMP-Leitfaden Annex 1: Herstellung steriler Arzneimittel
Im Kapitel „Überwachung der Reinräume und Reinluftanlagen“ des Annex 1 wird darauf hingewiesen, dass die Überwachungspunkte auf einer formalen Risikoanalyse basieren sollten (Quelle: EU-GMP-Leitfaden Annex 1).
Zur Vorgehensweise bei der Klassifizierung wird auf die EN ISO 14644-1 verwiesen mit dem Hinweis, dass die Methodik bei der Klassifizierung deutlich von der Überwachung der Umgebung im Betriebszustand abzugrenzen sei.
Bei der Auswahl des Überwachungssystems sollen außerdem alle gegenwärtigen Risiken der bei den Herstellungsschritten verwendeten Stoffe in Betracht gezogen werden.
Bei aseptischen Herstellungsvorgängen sollen häufige Kontrollen in Form von volumetrischen Luft- und Oberflächenproben durchgeführt werden (z. B. Tupfer und Kontaktplatten). Die Probenahmeverfahren während der Arbeit dürfen den Schutz der Bereiche nicht beeinträchtigen.
Das Kapitel „Fertigstellung steriler Produkte“ enthält den Hinweis, dass partiell verschlossene Behältnisse (z. B. beim Gefriertrocknen) so lange unter Klasse-A-Bedingungen gehalten werden sollen, bis der Stopfen vollständig eingeführt ist.
Dieser Punkt ist für gefriergetrocknete Produkte von Interesse, die nach der Abfüllung noch in die Gefriertrocknungsanlagen geladen werden müssen, wie es im hier geschilderten Beispiel der Fall ist.
2.2 PIC/S Recommendation on the Validation of Aseptic Processes
Laut Kapitel 7 „Environmental and Personnel Monitoring“ sollte im Rahmen der mikrobiellen und Partikelüberwachung der für die Überwachung gewählte Ort überprüft werden, um sicherzustellen, dass die Positionen den Worst Case widerspiegeln. Für die Raumüberwachung sollte das Monitoring an Stellen durchgeführt werden, an denen die meiste Bedieneraktivität herrscht. Für die Überwachung des Befüllungsprozesses sollte das Monitoring in der Nähe des Befüllungsbereichs und an Stellen durchgeführt werden, an denen die Messsonden so exponiert sind, dass eine Bedieneraktivität innerhalb dieser Bereiche erfasst werden kann. Eine Überwachung mit Probenahmesonden, die so angebracht sind, dass sie die Luft aus dem High-Efficient-Particulate-Air(HEPA)-Filter und nicht die Luft in unmittelbarer Umgebung der kritischen Zonen erfasst wird, sollte vermieden werden. Die Position der Probenahmevorrichtung sollte jedoch die Laminarität des Luftstroms in der kritischen Zone nicht beeinträchtigen. Im Rahmen der initialen Validierung sollte überprüft werden, dass die Worst-Case-Positionen korrekt identifiziert wurden. Diese können während der Prozesssimulationstests erneut bestätigt werden.
Die mikrobielle Überwachung sollte ebenfalls in Bereichen mit hoher Bedieneraktivität durchgeführt werden. Es ist nicht ungewöhnlich, dass Absetzplatten und Luftprobenstellen weit entfernt von solchen Bereichen platziert sind. Ein typisches Beispiel ist, dass die Sedimentationsplatten weit hinten an der Abfüllmaschine angebracht sind, wo es wenig oder keine Bedieneraktivität gibt.
Das Gleiche gilt für die Luftprobenahme. Es ist daher wichtig, die Aktivität des Bedieners über einen bestimmten Zeitraum hinweg zu beobachten und sicherzustellen, dass die Messstellen so platziert werden, dass der Einfluss der Bedieneraktivität überwacht werden kann (Quelle: PIC/S Recommendation on the Validation of Aseptic Processes).
2.3 DIN EN ISO 14644-1
Die Norm „Reinräume und zugehörige Reinraumbereiche – Teil 1: Klassifizierung der Luftreinheit anhand der Partikelkonzentration – EN ISO 14644-1:2015“ beschreibt die Vorgehensweise bei der Klassifizierung von Reinräumen.
Im Anhang A „Referenzverfahren zur Bestimmung der Klassifizierung der Luftreinheit anhand der Partikelkonzentration“ kann die Mindestanzahl von Probenahmeorten einer Tabelle entnommen werden (Abb. 1).
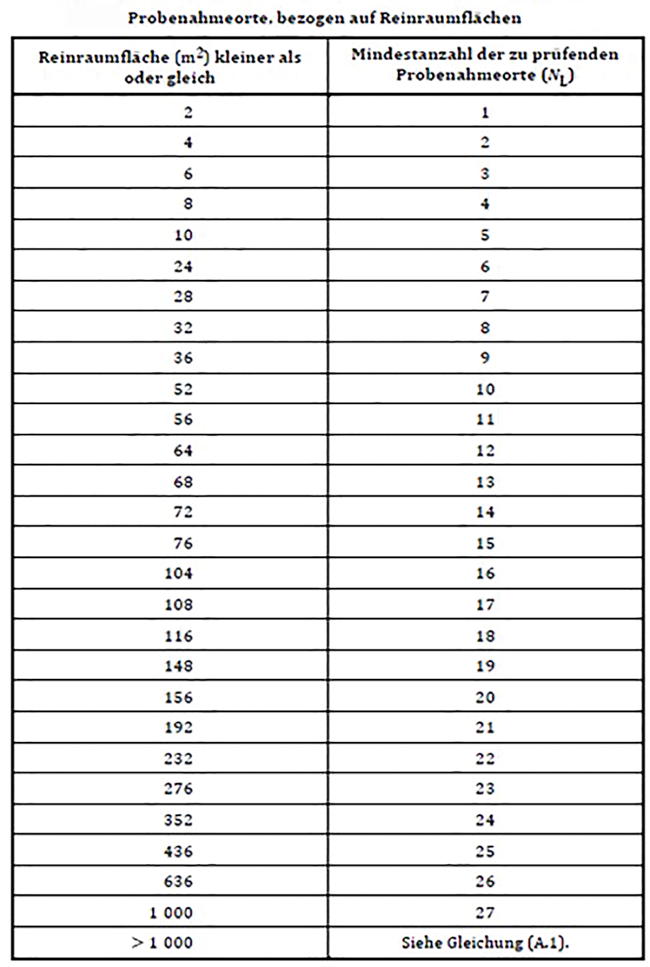
Für die Anordnung der Probenahmeorte gelten laut Norm folgende Regeln:
Ermittlung der Mindestanzahl der Probenahmeorte
Unterteilung des gesamten Raumes in gleich große NL-Abschnitte
Innerhalb jedes Abschnitts ist ein Probenahmeort auszuwählen, der als repräsentativ für die Eigenschaften des Abschnitts betrachtet wird.
An jedem Probenahmeort ist die Sonde des Partikelzählers auf der Höhe der Arbeitsaktivität oder an einem anderen festgelegten Punkt anzuordnen.
Für Bereiche, die als kritisch eingestuft wurden, können zusätzliche Messstellen festgelegt werden. In Bereichen mit turbulenzarmer Verdrängungsströmung sind Probenahmeorte, welche direkt unterhalb der nicht-diffusen Zuluftquellen angeordnet sind, nicht repräsentativ. Die Probenahmesonde muss dabei punktuell im Luftstrom positioniert sein. Wenn die Richtung des zu prüfenden Luftstromes nicht zu regeln oder voraussagbar ist (z. B. turbulente Verdünnungsströmung), dann muss der Einlass der Probenahmesonde senkrecht nach oben gerichtet sein (Quelle: DIN EN ISO 14644-1).
2.4 DIN EN ISO 14644-2
Die Norm „Reinräume und zugehörige Reinraumbereiche – Teil 2: Überwachung zum Nachweis der Reinraumleistung bzgl. Luftreinheit anhand der Partikelkonzentration – EN ISO 14644-2:2015“ beschreibt die Maßnahmen zur Überwachung von Reinräumen.
In Kapitel 4 „Erstellung, Umsetzung und Aufrechterhaltung eines Überwachungsplans“ werden die Notwendigkeit eines Plans hervorgehoben sowie die wichtigsten Aspekte bei dessen Erstellung und Umsetzung spezifiziert:
Anwendung geeigneter Instrumente zur Risikobewertung, um das Risiko von nachteiligen Kontaminationsereignissen zu verstehen, zu bewerten und zu dokumentieren
Entwicklung eines schriftlichen Überwachungsplans
Prüfen und Genehmigen des Plans
Umsetzen des Plans mittels Durchführung der Überwachung
Analysieren der von den Überwachungsmaßnahmen abgeleiteten Daten, Durchführen einer Trendanalyse, wenn zutreffend, und Dokumentieren der Leistung
Umsetzen und Dokumentieren der Maßnahmen oder der erforderlichen Korrekturmaßnahmen
Durchführen einer regelmäßigen Überprüfung/Bewertung des Überwachungsplans
Im Folgenden wird die Notwendigkeit der Risikobewertung bei der Erstellung des Überwachungsplans begründet. Die Risikobewertung muss erfolgen:
um einen Überwachungsplan durch die Bestimmung von Faktoren zu erarbeiten, die die Leistungsmerkmale der Anlage hinsichtlich der Aufrechterhaltung der vereinbarten Luftreinheit anhand der Partikelkonzentration des Reinraums oder reinen Bereichs beeinflussen können, und
um die Anforderungen an die Überwachung festzulegen, um den Leistungsnachweis bereitzustellen.
An den Überwachungsplan selbst werden folgende Anforderungen gestellt:
Der Überwachungsplan muss eine Auflistung und Begründung aller zu überwachenden Parameter enthalten, einschließlich derjenigen, die die Konzentration der luftgetragenen Partikel beeinflussen können.
Der Überwachungsplan muss das Ergebnis der Risikobewertung berücksichtigen.
Der Überwachungsplan muss eine Beschreibung und Begründung der Messverfahren enthalten.
Der Überwachungsplan muss Genauigkeit, Instandhaltung und Kalibrierung der Messgeräteausrüstung für die Überwachung berücksichtigen.
Im Rahmen des Überwachungsplans sind die ausgewählten Überwachungsorte zu identifizieren und zu begründen. Die Überwachungsorte müssen in 3 Dimensionen definiert sein.
Bei der Entwicklung des Überwachungsplans sind gemäß Anhang A, Kapitel A.4 „Überwachungssystem der luftgetragenen Partikel“ folgende Aspekte zu berücksichtigen:
Die Konfiguration des Überwachungssystems beruht auf der Bewertung der folgenden Systemmerkmale:
Wirksamkeit der Sammlung luftgetragener Partikel
Eignung des Systems für die Überwachung der ausgewählten Partikelgröße(n)
Zugänglichkeit für Instandhaltung, Kalibrierung und Reparatur
Die Konfigurierung und Ausrichtung der Probenahmesonde muss unter Berücksichtigung der Luftströmung (z. B. isokinetisch oder anisokinetisch) erfolgen.
Anmerkung
Es kann unzweckmäßig sein, eine Probenahmesonde direkt unter einem Zuluftdurchlass mit einem Schwebstofffilter (HEPA-Filter) in einer Konfiguration mit turbulenter Verdünnungsströmung anzuordnen, da so ein Ort für den Reinraum oder reinen Bereich nicht repräsentativ sein kann und den Nachweis von Kontaminationen während des Betriebs verhindern kann.
Die mögliche nachteilige Auswirkung des Probenahmesystems auf das Verfahren oder die Verfahrensumgebung sollte berücksichtigt werden (z. B. mögliche Auswirkungen der Entnahmerate des Probenvolumens in kleinen Volumen) (Quelle: DIN EN ISO 14644-2).
2.5 VDI 2083: Blatt 3.1
In den VDI-Richtlinien zu „Reinraumtechnik – Messtechnik in der Reinraumluft – Monitoring“ wird in Kapitel 1 „Anwendungsbereich“ eine klare Differenzierung zwischen Klassifizierungsmessungen und Monitoring vorgenommen.
Das Monitoring (Überwachung von Reinräumen) muss nicht nach DIN EN ISO 14644-1 vorgenommen werden. Es darf mit einer kleineren Anzahl von Messpunkten und einem kleineren Messvolumen durchgeführt werden. Eine formelle Risikoanalyse der Monitoringdaten über einen angemessenen Betriebszeitraum hinweg sollte als Grundlage für die Festlegung von Messhäufigkeiten, Messorten und Grenzwerten dienen. Messhäufigkeiten, Messorte und Grenzwerte sollten sich am Prozess orientieren.
In Kapitel 6 „Partikelmonitoring“ wird die Vorgehensweise näher spezifiziert. Das Design eines Partikelmonitoringsystems hängt ab vom Einsatzbereich, von den gesetzlichen Bestimmungen, Sicherheitsaspekten und den Wünschen des Nutzers nach Übersichtlichkeit und Komfort. Die Probenluft wird an der Messstelle entnommen und mittels Trichter möglichst schonend auf die Transportgeschwindigkeit von ca. 15 m/s (im Probenahmeschlauch) gebracht. Die Probenahme im turbulenzarmen Luftstrom geschieht mittels einer isokinetischen Probenahmevorrichtung.
Die an der Messstelle verwendeten Materialien sollen korrosionsbeständig, abriebfest und von hoher Oberflächengüte sein.
Anzahl und Lage der Messstellen sollen über eine Risikoanalyse bestimmt und definiert werden. Messstellen sollen, soweit nicht anderweitig festgelegt, repräsentativ für den zu beurteilenden Reinraumbereich angeordnet werden. Die Entfernung der Probenahme vom kritischen Bereich muss so gewählt werden, dass die Sonde keinen Einfluss auf den Zustand des kritischen Bereichs hat. Die Messungen müssen trotzdem die Bedingungen im reinen Bereich widerspiegeln. Bei der Sterilfertigung in der pharmazeutischen Industrie sollte die Messstelle nicht weiter als 30 cm von dem für das Produkt kritischen Punkt entfernt sein.
Im Unterpunkt „Mikrobiologisches Monitoring“ des Kapitels „Mikrobiologisches Monitoring“ wird ein schriftlich dokumentierter Probenahmeplan gefordert. Dieser soll gemäß den Kriterien einer Risikoanalyse, den kritischen Messstellen und der Häufigkeit der Kontrollen ausgearbeitet werden. Pro Raum >300 m3 sollten mindestens 3 Messstellen bestimmt werden (Quelle: VDI 2083: Blatt 3.1).
2.6 DIN EN ISO 14698-1
Nach der Norm „Reinräume und zugehörige Reinraumbereiche – Biokontaminationskontrolle – Teil 1: Allgemeine Grundlagen – EN ISO 14698-1:2003“ sollen laut Kapitel 5.3.2.6 „Probenahmeorte“ die Messstellen für die mikrobiologische Überwachung entsprechend dem gewählten betrieblichen System festgelegt werden und in den Probenahmeplan aufgenommen werden. Es können mehr als eine Probe an einer Stelle bzw. eine unterschiedliche Anzahl von Proben an verschiedenen Stellen genommen werden. Die Probenahme muss an den in einem schriftlich niedergelegten Ablauf festgelegten mikrobiologischen Kontrollpunkten durchgeführt werden.
2.7 ZLG Aide-mémoire 07120606
In „Überwachung von Sterilherstellern“ wird in Kapitel 5 „Überwachung der Reinräume und Reinluftanlagen“ die Partikel- und Keimzahlmessung behandelt. Bei Monitoring in Klasse-A-Bereichen sollen die Partikelmessstellen basierend auf einer Risikoanalyse produktnah angeordnet werden. Abgesehen von der kontinuierlichen Messung am Abfüllpunkt kann auf Basis einer Risikobewertung an weiteren Probenahmestellen im Klasse-A-Bereich eine kontinuierliche Partikelmessung erforderlich sein.
Art und Umfang des mikrobiologischen Monitorings beruhen ebenfalls auf einer Risikobewertung. Bei Neuanlagen oder wesentlichen Änderungen beginnt das Monitoring mit einem umfangreichen engmaschigen Basismonitoring mit vielen Messpunkten. Bei der Auswahl der geeigneten Messpunkte hat sich das Verfahren des Mappings bewährt.
Grundsätzlich wird der Einsatz von Luftkeimmessungen, Sedimentationsplatten, Kontaktplatten und Handschuhabdruck (5-Finger-Print) erwartet. Nach kritischen Arbeitsgängen müssen mittels geeigneter Verfahren mikrobiologische Abklatschkontrollen des Personals und der Oberflächen durchgeführt werden.
Alle Messpunkte müssen genau beschrieben sein; ein Lageplan wird erwartet. Ein rollierendes System der Probenahmestellen im Bereich der kritischen Herstellungsschritte (RRK-A- und RRK-B-Bereiche; insbesondere aseptische Abfüllung, Bearbeitung des offenen Produktes) ist abzulehnen.
Die verwendeten Methoden sind im Rahmen der Risikobewertung in Abhängigkeit des Prozesses festzulegen, z. B. kann die aktive Luftkeimmessung gegenüber der Sedimentationsplatte aussagekräftigere Ergebnisse erzielen.
Die Probenahmesonde für die Luftkeimsammlung muss punktuell im Luftstrom positioniert sein. Sie ist grundsätzlich in unmittelbarer Nähe zu der Stelle mit der höchsten und kritischsten Herstellungsaktivität zu positionieren.
Frequenz, Lage und Anzahl der Monitoringmesspunkte basieren auf einer formalen Risikoanalyse, den Ergebnissen der Reinraumklassifizierung und des o. g. Basismonitorings. Alle kritischen Bereiche müssen erfasst werden.
2.8 FDA Guidance for Industry Sterile Drug Products Produced by Aseptic Processing
Der „Guide to Aseptic Processing“ der FDA empfiehlt in Kapitel IV „Buildings and Facilities“, dass Messungen zur Bestätigung der Luftreinheit in kritischen Bereichen an den Stellen vorgenommen werden, an denen das größte potenzielle Risiko für das exponierte sterilisierte Produkt, die Behälter und Verschlüsse besteht. Die Partikelzählsonde sollte in einer Ausrichtung platziert werden, die nachweislich eine aussagekräftige Probe ergibt. Eine regelmäßige Überwachung sollte während jeder Fertigungsschicht durchgeführt werden. Hierbei wird empfohlen, die Überwachung nicht lebensfähiger Partikel mit einem Fernzählsystem durchzuführen. Diese Systeme sind in der Lage, umfassendere Daten zu sammeln, und sind im Allgemeinen weniger invasiv als tragbare Partikelzähler.
Laut dem Kapitel „Laboratory Controls“ ist es wichtig, dass die Stellen, die das größte mikrobiologische Risiko für das Produkt darstellen, ein wesentlicher Bestandteil des Monitorings sind. Es ist besonders wichtig, die mikrobiologische Qualität der kritischen Bereiche zu überwachen, um festzustellen, ob während der Abfüll- und Verschließvorgänge die aseptischen Bedingungen eingehalten werden oder nicht. Luft- und Oberflächenproben sollten an den Stellen entnommen werden, an denen während der Produktion eine signifikante Aktivität oder Produktexposition stattfindet.
Alle Umweltüberwachungsstandorte sollten in Standard Operating Procedures (SOPs) mit ausreichenden Details beschrieben werden, um eine reproduzierbare Probenahme an einem bestimmten untersuchten Standort zu ermöglichen. Schriftliche SOPs sollten auch Elemente wie
Häufigkeit der Probenahme
Zeitpunkt der Probenahme (d. h. während oder nach Abschluss des Betriebs)
Dauer der Probenahme
Probenumfang (z. B. Fläche, Luftvolumen)
spezifische Probenahmegeräte und -techniken
Alarm- und Aktionsstufen sowie
angemessene Reaktion auf Abweichungen von Alarm- oder Aktionsstufen behandeln.
Bei der Positionierung der mikrobiologischen Überwachungssonden sollte der Bezug des Probenahmeortes zur betrieblichen Aktivität hergestellt werden. Die Messstellen sollten auf der Notwendigkeit beruhen, eine angemessene mikrobiologische Kontrolle in der gesamten sterilen Produktionsanlage aufrechtzuerhalten. Bei der Festlegung der Überwachungsorte sollten auch historische Daten des Umgebungsmonitorings, Medienbefüllungen, Reinraumqualifizierungen und Desinfektionsstudien berücksichtigt werden. Daten aus ähnlichen Prozessen können ebenfalls hilfreich sein, um Aktions- und Alarmstufen festzulegen, insbesondere für neue Prozesse und Anlagen.
Die Bewertung der mikrobiellen Qualität der Luft sollte die Verwendung aktiver Geräte beinhalten, einschließlich, aber nicht beschränkt auf Impaktions-, Zentrifugal- und Membran- (oder Gelatine-)Probenehmer. Jedes Gerät hat bestimmte Vor- und Nachteile, obwohl alle die Prüfung der Anzahl von Organismen pro Volumen der beprobten Luft ermöglichen.
Eine weitere Methode ist die Verwendung von passiven Luftprobennehmern, z. B. Sedimentationsplatten (Petrischalen mit Nährmedium, die der Umgebung ausgesetzt werden). Da nur Mikroorganismen nachgewiesen werden, die sich auf der Agaroberfläche absetzen, können Sedimentationsplatten als qualitative oder halbquantitative Luftüberwachungsgeräte verwendet werden. Ihr Wert in kritischen Bereichen wird dadurch erhöht, dass die Platten an Stellen positioniert werden, die das größte Risiko einer Produktkontamination darstellen. Die durch passive Luftprobenahme erzeugten Daten können nützlich sein, wenn sie in Kombination mit den Ergebnissen anderer Arten von Luftproben betrachtet werden.
2.9 Zusammenfassung der Kernaussagen
Um die Compliance in RRK-A-Bereichen sicherzustellen, müssen die Partikel- und Keimzähler korrekt positioniert werden. Die Auswahl der Positionen erfolgt basierend auf einer prozessorientierten Risikobewertung. Die aus der Risikoanalyse resultierenden Messstellen für Partikel und Keime müssen im Rahmen der Erstellung eines Überwachungsplans festgelegt und begründet sein. Die Probenahmestellen sind so festzulegen, dass alle kritischen Bereiche (Worst Case) abgedeckt sind. Es ist ein Lageplan zu erstellen, in welchem die Messpositionen in 3 Dimensionen definiert sind. Der Messort ist so zu wählen, dass die Messsonde nicht weiter als 30 cm von dem für das Produkt kritischen Punkt entfernt ist und die Höhe der Messsonde der Höhe der Arbeitsaktivität entspricht. Die Messverfahren dürfen den Schutz der Bereiche nicht beeinträchtigen.
Repräsentative Positionen sind z. B. Orte mit hoher Operatoraktivität. Bei Abfüllprozessen muss eine kontinuierliche Partikelmessung am Abfüllpunkt nahe des Abfüllbereichs stattfinden. Die Messstelle ist so zu wählen, dass ein potenzieller Einfluss der Operatoraktivität erfasst werden kann.
Beim Monitoring der mikrobiologischen Aktivität ist ebenfalls der Einfluss der Operatoraktivität zu berücksichtigen. Die Bereiche mit hoher Operatoraktivität sind zu identifizieren und die Messsonden entsprechend zu platzieren.
3. Definition der Anforderung im Rahmen der URS
Der EU-GMP-Leitfaden – Anhang 15: Qualifizierung und Validierung beschreibt seine Anforderungen in Bezug auf die Benutzeranforderung (URS) in folgender Weise:
„Die Spezifikation für Ausrüstung, Einrichtungen, Betriebsmittel oder Systeme sollte in Form einer URS und/oder einer Funktionsspezifikation erfolgen. Die wesentlichen Qualitätselemente müssen zu diesem Zeitpunkt integriert und eventuelle GMP-Risiken auf ein akzeptables Niveau reduziert werden. Die URS sollte während des gesamten Validierungslebenszyklus als Bezugspunkt dienen.“ (Quelle: EU-GMP-Leitfaden – Anhang 15)
Ziel ist die vollständige und korrekte Definition aller Anforderungen des Auftraggebers hinsichtlich Liefer- und Leistungsumfang. Nachträgliche Anforderungen oder Änderungen führen zu folgenden Risiken:
steigender Aufwand
steigende Kosten
Terminverzug
Somit müssen die regulatorischen Anforderungen nun im Rahmen der URS umgesetzt werden. Zusätzlich müssen weitere Aspekte des Umgebungsmonitorings berücksichtigt werden, die einen Einfluss auf die Konstruktion und Auslegung der Anlage haben.
Hierzu zählen:
Aufzeichnung der Partikelmonitoringdaten
Alarmierung bei Abweichungen (Warn-/Alarmgrenzen)
Dokumentation der Durchführung der aktiven Luftkeimmessung
automatischer Start der Partikelmessung nach der Dekontamination des Isolators
Zugänglichkeit/Erreichbarkeit der Probenahmestellen über Handschuheingriffe
Die Anforderungen können im Rahmen der URS wie folgt formuliert werden:
Es sind je ein Partikelmonitoring und eine Luftkeimzahlbestimmung im Isolator auf der Füllmaschine entsprechend den Anforderungen der Guidelines vorzusehen.
Anzahl und Positionierung müssen den Anforderungen gemäß EU-GMP-Leitfaden und FDA Aseptic Guide entsprechen.
Die Festlegung der Anzahl und Position muss auf Basis einer formalen Risikoanalyse beruhen und im Rahmen des Design-Review und Mock-up bestätigt werden.
Es ist ein Partikelmonitoringsystem zur Erfassung des folgenden Grenzwertes vorzusehen: 100 Partikel/ft3
Es ist ein System zur aktiven Luftkeimzahlbestimmung im Isolator vorzusehen. Messung der Keimzahl über MAS 100 (<1 KBE/m3) oder vergleichbares System.
Es sind Ablagevorrichtungen für Sedimentationsplatten vorzusehen: Grenzwert <1 KBE/4 h.
Es sind Aufhänge- und Ablagevorrichtungen für das Oberflächenmonitoring vorzusehen. 1 KBE/25 cm2 Warngrenze, 3 KBE/25 cm2 Aktionsgrenze.
Positionierung von Messungen zur Keimzahlbestimmung im Bereich der Packmittelzuführungen
Beachtung strömungsgünstiger Geometrien der Funktionsbaugruppen
Platzreserve für Messeinrichtungen in der Maschine
Im Isolator steht ausreichend Platz zur Bevorratung von mikrobiologischem Probematerial, Hilfsmitteln und Werkzeugen zur Verfügung. Entsprechende Aufhänge- und Ablagevorrichtungen sind vorzusehen.
Gute Zugänglichkeit zu allen relevanten Punkten über die Handschuheingriffe; die Vorrichtungen zum Ausschleusen der Proben (Sedimentationsplatten, Luftkeimproben) sind gut über die Handschuheingriffe erreichbar.
Status der Anlage: Produktionsvorbereitung/Stand-by. Alle Monitoringeinrichtungen sind aktiv, die überwachten Parameter werden gemäß Sollwerten gefahren. Der Status der Anlage ist steril.
Status der Anlage: Produktion. Alle Monitoringeinrichtungen sind aktiv, die überwachten Parameter werden gemäß Sollwerten gefahren. Der Status der Anlage ist steril. Die Parameter werden überwacht und geregelt.
4. Risikobasierte Festlegung der Monitoringpunkte
Voraussetzung für die Durchführung der risikobasierten Festlegung der Monitoringpunkte ist, dass das Layout der Abfüllanlage und die Prozessabläufe bekannt sind. In diesem Fallbeispiel wird eine Abfüllanlage mit RABS betrachtet, welche Produkte für eine anschließende Gefriertrocknung abfüllt.
Das Risiko für das sterile Produkt ist am größten bei Prozessschritten, bei denen die Flaschen nicht vollständig verschlossen sind, d. h. beim Abfüllen, beim Transport zu den Gefriertrocknern, beim Be- und Entladen der Gefriertrockner, beim Transport zu den Lagerwagen, bei der Zwischenlagerung und beim Transport zur Verschließmaschine. Das Risiko einer Partikelkontamination ist in der Nähe von beweglichen Teilen wie Drehtischen, Füllern, Stopfern, Transportwagenregalen usw. hoch. Ein erhöhtes Risiko für eine Kontamination mit Mikroorganismen ist immer dann gegeben, wenn ein Eingriff durch einen Bediener erforderlich ist.
Grundlage für die Bewertung sind der Produktfluss, Prozessabläufe und mögliche manuelle Tätigkeiten bei der Befüllung und Beladung der Gefriertrockner. Folgende Kriterien werden für die Bewertung des Risikos bzw. für die Auswahl der Messstellen herangezogen:
potenzielle Partikelfreisetzung durch Reibung des Primärpackmittels
potenzielle Partikelfreisetzung durch bewegte Anlagenteile
Übergang (Schnittstelle) von verschiedenen Reinheitsklassen
Einhaltung der behördlichen Anforderungen
offene Primärverpackung vorhanden
manueller Eingriff
offenes Produkt vorhanden
Arbeitshöhe
Flaschengrößen
Das Vorgehen zur risikobasierten Festlegung der Monitoringpunkte wird im Folgenden an einem Fallbeispiel dargestellt.
4.1 Prozessablauf/Fallbeispiel
Produkttanks werden an die Abfüllanlage zur Inline-Filtration und anschließenden Abfüllung angeschlossen. Die Flaschen werden aus dem Heißlufttunnel in die Abfüllanlage transportiert, wo sie in RRK A verarbeitet werden. Die Flaschen werden zur Füllstation transportiert und dort befüllt. Nach dem Füllen werden die Flaschen für die anschließende Gefriertrocknung (GT) teilverschlossen und zum Magazinierer transportiert. Dort werden die Flaschen zu einem Paket eingerahmt und zur Übergabeposition geschoben.
Der Transferwagen wird an den Magazinierer angedockt und das Flaschenpaket übergeschoben. Der Transferwagen wird zur Gefriertrocknungsanlage geschoben und das Flaschenpaket dort in die GT eingeschoben.
4.2 Produktfluss/Materialfluss
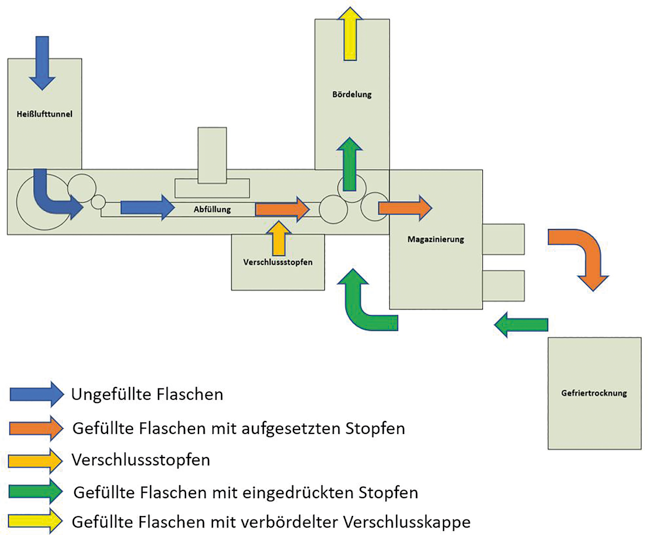
Die Darstellung der Produkt- und Materialflüsse (Abb. 2) fließt in die spätere Risikobewertung ein. In dieser Darstellung sind die Materialwege der Primärpackmittel und der Produktfluss der teilverstopften und auch verstopften Flaschen ersichtlich. Jeder dieser Flusswege hat ein eigenes Risikopotenzial, was berücksichtigt werden muss.
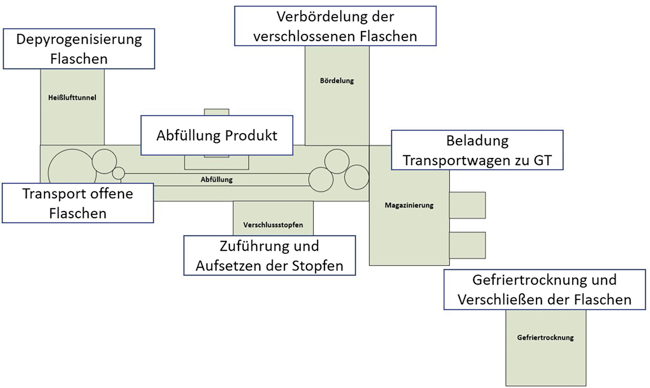
4.3 Prozessabläufe
Die Darstellung der Prozessabläufe (Abb. 3) fließt auch in die spätere Risikobewertung ein. Hier sind die einzelnen Prozesse im Rahmen der Abfüllung ersichtlich. Jeder einzelne Prozess hat ein eigenes Risikopotenzial bzgl. der Kontamination des Produktes mit Partikeln oder Mikroorganismen. Diese unterschiedlichen Risikopotenziale müssen im Rahmen der Risikobewertung entsprechend berücksichtigt werden und führen zu entsprechenden Maßnahmen.
4.4 Risikoanalyse/Risikobewertung
Ein dokumentiertes, transparentes und reproduzierbares Verfahren sollte zur Durchführung der Risikobewertung genutzt werden. Hierzu bietet sich die Risikoanalyse an. In die Bewertung der Risikoanalyse fließen die Erkenntnisse aus der Betrachtung der Produktflüsse/Materialflüsse, Prozessabläufe und den manuellen Tätigkeiten ein. Diese Bewertung ist für einige ausgewählte Punkte in der Tab. 1 dargestellt.
Tabelle 1
Position/Prozess | Risiko | Maßnahme |
Position: Abfüllmaschine nach Heißlufttunnel Prozess: Transport offener Primärverpackungen Flaschenformat: 50 ml | Allgemeine Risikofaktoren: – hohe Verweildauer der geöffneten Fläschchen – hohe Anzahl geöffneter Fläschchen – offenes Primärverpackungsmaterial – große Aufprallfläche (Drehteller) Risikopotenzial Partikel: – mögliche Partikelfreisetzung durch sich bewegende Maschinenteile und die Reibung des Primärverpackungsmaterials Risikopotenzial Mikrobiologie: – Luft strömt aus den umliegenden Gebieten ein (Tunnel) – manuelles Eingreifen bei Störungen | Erforderliche Überwachungspositionen: – 1 mikrobielle Sammlung – 1 Partikelsammlung Höhe der Arbeitsaktivität: Flaschenöffnung |
Position: Abfüllmaschine/ Abfüllung Prozess: – Transport offener Primärverpackungen – Füllen der geöffneten Primärverpackungen Flaschenformat: 50 ml | Risikopotenzial Partikel: – mögliche Partikelfreisetzung durch bewegliche Maschinenteile oberhalb der Flaschenöffnung Risikopotenzial Mikrobiologie: – in der Nähe des offenen Produkts | Erforderliche Überwachungspositionen: – 1 mikrobielle Sammlung – 1 Partikelsammlung Höhe der Arbeitsaktivität: Flaschenöffnung |
Position: Stopfenzuführung Prozess: Auspacken und Zuführung der Stopfen Flaschenformat: 50 ml | Risikopotenzial Partikel: – mögliche Freisetzung von Partikeln durch das Auspacken und Zuführen von Stopfen Risikopotenzial Mikrobiologie: – Luft strömt aus den umliegenden Gebieten ein – Bedienereingriff beim Auspacken und Zuführen der Stopfen | Erforderliche Überwachungspositionen: – 1 mikrobielle Sammlung – 1 Partikelsammlung Höhe der Arbeitsaktivität: Stopfenbunker |
Position: Stopfensortierung nach Stopfenzuführung Prozess: – Sortierung der Stopfen Flaschenformat: 50 ml | Risikopotenzial Partikel: – mögliche Partikelfreisetzung durch das Sortieren von Stopfen Risikopotenzial Mikrobiologie: – Luft strömt aus den umliegenden Gebieten ein – Bedienereingriff beim Auspacken und Zuführen der Stopfen | Erforderliche Überwachungspositionen: – 1 mikrobielle Sammlung – 1 Partikelsammlung Höhe der Arbeitsaktivität: Sortiertopf |
Position: Stopfensetzer Transportstrecke zum Magazinierer Prozess: – Aufsetzen der Stopfen – Transport der teilverstopften Flaschen Flaschenformat: 50 ml | Risikopotenzial Partikel: – mögliche Partikelfreisetzung durch bewegliche Maschinenteile oberhalb der Flaschenöffnung Risikopotenzial Mikrobiologie: – in der Nähe des offenen Produkts | Erforderliche Überwachungspositionen: – 1 mikrobielle Sammlung – 1 Partikelsammlung Höhe der Arbeitsaktivität: Flaschenöffnung |
4.5 Messpunkteplan
Aus der durchgeführten Risikobewertung und unter Beachtung der folgenden regulatorischen Anforderungen ergibt sich der Messpunkteplan, welcher in Abb. 4 zu sehen ist.
Der Messort ist so zu wählen, dass die Messsonde nicht weiter als 30 cm von dem für das Produkt kritischen Punkt entfernt ist und die Höhe der Messsonde der Höhe der Arbeitsaktivität entspricht.
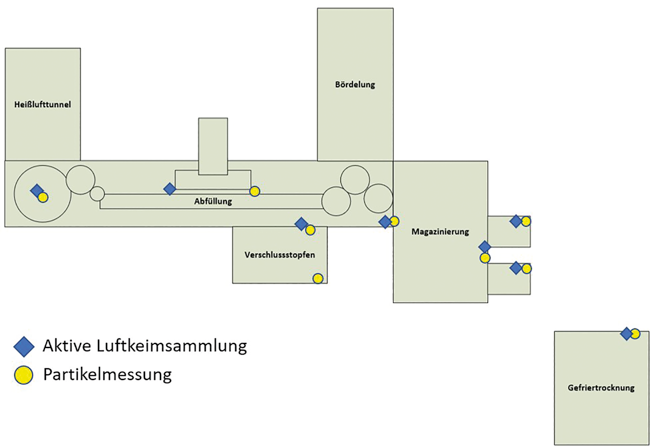
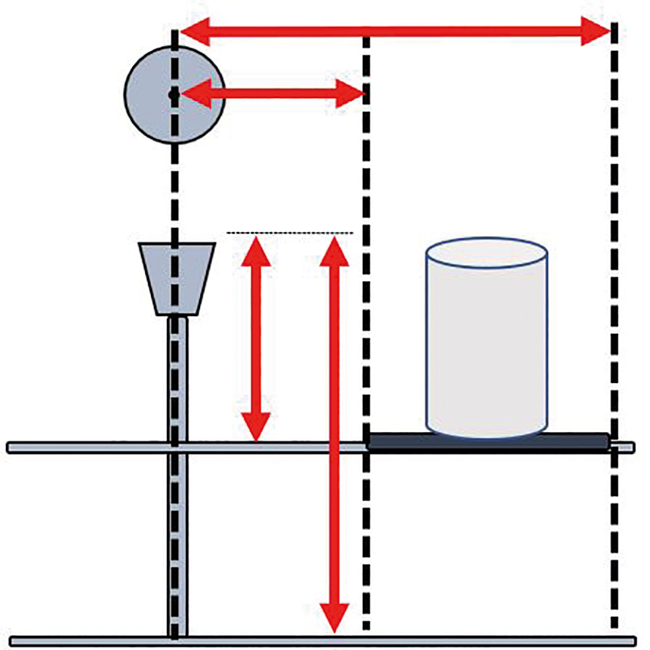
Es ist ein Lageplan zu erstellen, in welchem die Messpositionen in 3 Dimensionen definiert sind. Ein solcher Lageplan ist für eine Position einer Partikelsonde beispielhaft in Abb. 5 dargestellt. Das größte Flaschenformat zur Festlegung der Höhe der Arbeitsaktivität (Flaschenöffnung) wurde berücksichtigt.
5. Verifizierung der Monitoringpunkte in der Design-Phase
Die Phasen im Lebenszyklus einer Anlage lassen sich in die folgenden Abschnitte einteilen:
Spezifikation/Lastenheft
Planung/Design-Phase
Installation/Inbetriebnahme
Qualifizierung/Validierung
Betrieb/Wartung
Änderungen/Re-Qualifizierung-Validierung
Außerbetriebnahme
Die Festlegung der Anzahl und der Position der Monitoringpunkte muss im Rahmen der Planung/Design-Phase erfolgen, da diese Messstellen ein fester Bestandteil der Anlage sind. Eine Änderung im Rahmen der Installation und Inbetriebnahme lässt sich hier i. d. R. nicht ohne große Aufwände realisieren.
5.1 Design-Review/Design-FMEA
Zur Bestätigung der Anzahl und Position der Monitoringpunkte bietet sich die Designqualifizierung (DQ) an. Denn diese ist der erste Schritt der Qualifizierung der Ausrüstung. Im Rahmen der DQ wird die Übereinstimmung des Designs mit den GMP-Anforderungen nachgewiesen und dokumentiert. Zur Dokumentation der Überprüfung sollte ein Design-Review durchgeführt werden, welches eine umfassende und systematische Untersuchung und Bewertung darstellt. Hierzu sollte der systematische Ansatz einer Design-FMEA (Abb. 6) angewendet werden, die darauf abzielt, Fehler schon in der Planungs- und Konstruktionsphase zu erkennen und zu vermeiden. Durch die Umsetzung der hieraus abgeleiteten Maßnahmen soll die Design-FMEA zur Erhöhung der Systemsicherheit, -zuverlässigkeit und -verfügbarkeit beitragen. Im Rahmen der Design-FMEA wird die finale Konstruktion gegen die Vorgaben aus dem Messpunkteplan abgeglichen. Hierbei werden die folgenden Punkte bewertet:
Position vorhanden
Position korrekt (Abstand zum Produkt/Höhe)
Position zugänglich für manuelle Eingriffe

5.2 Mock-up
Im Rahmen der Design-FMEA findet die erste Bestätigung der Position der Messstellen statt. Im Rahmen der Mock-up-Studie sollen nun die folgenden Punkte verifiziert werden:
Ergonomie/Bedienbarkeit (Handschuheingriffe)
ausreichendes Sichtfeld bei Eingriffen
Durchführbarkeit aller Prozesse (Rüsten, Monitoring, Störungsbeseitigung)
Um ein Mock-up-Modell für einen Isolator oder RABS herzustellen, sind folgende Punkte zu beachten:
abgeschlossene mechanische Konstruktion der Anlage
finalisiertes Maschinenlayout inkl. Position der Monitoringpunkte
bauliche Gegebenheiten am Aufstellungsort (umgebender Raum)
Schnittstellen zu den Medien
Schnittstellen zur Klimatechnik
Schnittstellen zu anderen Anlagen
Definition der Materialflüsse (in und aus Isolator/RABS)
Definition der Produktionsabläufe
Definition der Nebenprozesse (Monitoring, Reinigung, Formatwechsel usw.)
Wie in Abb. 7 zu sehen ist, sind die Monitoringpunkte für die aktive Luftkeimsammlung und Partikelmessung fester Bestandteil des Mock-up-Modells, um die notwendigen Eingriffe ausreichend realistisch abbilden zu können. Eine nicht ausreichende Durchführung führt zu dem Risiko, dass es im späteren Routineprozess zu Störungen kommt, da Positionen schlecht zu erreichen sind oder es keine ausreichende Sicht auf die durchgeführte Tätigkeit gibt. Aus diesem Grund sollte ein Drehbuch/Plan zur Durchführung der Mock-up-Studie erstellt werden. Dieses Drehbuch sollte die folgenden Inhalte umfassen:
Überprüfen der Bedienbarkeit und des ausreichenden Sichtfeldes und ggf. Anpassung der Monitoringpositionen – Partikel
Überprüfen der Bedienbarkeit und des ausreichenden Sichtfeldes und ggf. Anpassung der Monitoringpositionen – Mikrobiologie
Simulation aller Eingriffe für das mikrobiologische Monitoring (Routine, Abschluss-Monitoring und Media Fill)

5.3 Abschluss der Design-Phase
Sind Design-Review und Mock-up-Studie abgeschlossen und konnten die Messstellen in beiden Prozessen bestätigt werden, sollte der Messpunkteplan auf seinen finalen Stand angepasst werden. So wurden in der Design-Phase das Risiko bzgl. Abweichungen und notwendiger Änderungen in Bezug auf die Monitoringpositionen minimiert und das größtmögliche Maß an Sicherheit für die nachfolgenden Phasen im Lebenszyklus erzeugt.
Cleanroom-and-Processes 2022, Nr. 1, Seite 32