COMP – News | Beitrag aus pharmind 88 | Nr. 7 | Seite XVII-XVIII (2026)
07.08.2026Der Ausschuss für Orphan-Arzneimittel (Committee for Orphan Medicinal Products, COMP) ist ein wissenschaftliches Gremium der Europäischen Arzneimittel-Agentur (European Medicines Agency, EMA), das ...

GKV-Spargesetze: Konsequentes Ausblenden der Realität
07.08.2026Wir können nicht mehr ausgeben, als wir einnehmen – das ist die politische Begründung für Gesetze à la GKV-Beitragssatzstabilisierungsgesetz. Damit mag man eine treue Krämerseele überzeugen ...
Reformprogramm der Bundesregierung: Jetzt müssen alle Zukunftstechnologien profitieren
07.08.2026Mit ihrem Reformprogramm setzt die Bundesregierung wichtige Impulse für den Industriestandort Deutschland. Geplante Maßnahmen zum Bürokratieabbau, schnellere Genehmigungsverfahren und eine neue ...

Fresenius gibt FDA-Zulassung für Rituximab-Biosimilar für den US-Markt bekannt
07.08.2026Fresenius Kabi, eine Betreibergesellschaft von Fresenius, gab bekannt, dass die US-amerikanische Food and Drug Administration (FDA) ein von Dr. Reddy's Laboratories (DRL) entwickeltes ...
Führungswechsel bei ZAHORANSKY USA
07.08.2026Personelle Weichenstellung am US-Standort von ZAHORANSKY: Zum 1. Juli 2026 hat Steve Bellocchio die Funktion des Präsidenten der ZAHORANSKY USA Inc. übernommen. Mit seiner ausgeprägten ...
Mehdi Hireche ist Geschäftsführer von Air Liquide in Deutschland
07.08.2026Mehdi Hireche wurde mit Wirkung zum 1. Juli 2026 zum Geschäftsführer der Air Liquide Deutschland GmbH ernannt. In dieser Funktion verantwortet er die Geschäftsentwicklung sowie das nachhaltige ...
Christian Reichardt stärkt als Chief Technology Officer (CTO) die technologische Weiterentwicklung von Optima Pharma
06.08.2026Mit Christian Reichardt hat Optima Pharma zum 1. April 2026 die Position des Chief Technology Officer (CTO) neu besetzt. Der Ingenieur bringt mehr als 17 Jahre internationale Erfahrung in der ...
Chillventa 2026: Pfeiffer präsentiert Lösungen für zuverlässige Lecksuche in Kälteanlagen und Rechenzentren
06.08.2026Vom 13. bis 15. Oktober 2026 stellt Pfeiffer Vacuum+Fab Solutions auf der Chillventa in Nürnberg seine Lösungen für die industrielle Dichtheitsprüfung vor. Im Fokus an Stand 6-440 in Halle 6 ...
BioNTech ernennt Dr. Guido Oelkers zum neuen Vorstandsvorsitzenden
06.08.2026BioNTech SE gab bekannt, dass der Aufsichtsrat Dr. Guido Oelkers zum neuen Vorstandsvorsitzenden (Chief Executive Officer, „CEO“) der BioNTech SE ernannt hat, der sein Amt spätestens zum 1. ...
Eppendorf unterzeichnet Vertrag für neuen Produktionsstandort in Penang, Malaysia
07.08.2026Die Eppendorf Gruppe plant einen neuen Produktionsstandort in Penang, Malaysia. Mit der Unterzeichnung des Pachtvertrags für das Grundstück im Bandar Cassia Technology Park macht das Unternehmen ...
Mobile Device Management in der Pharmaindustrie
07.08.2026Die Pharmaindustrie zählt zu den am stärksten regulierten Branchen überhaupt. Produktionsprozesse müssen lückenlos dokumentiert, Qualitätsstandards konsequent eingehalten und sämtliche ...
Branchenpartner
meistgelesen

Beitrag aus der Ausgabe 1/2026 der Zeitschrift pharmind
Stabilitätsprüfung von Wirkstoffen und Arzneimitteln
Der Entwurf der neuen ICH-Leitlinie Q1
Stabilitätsuntersuchungen sind ein essenzieller Bestandteil der Arzneimittelentwicklung und -zulassung. Sie dienen dazu, die Qualität, Sicherheit und Wirksamkeit eines Arzneimittels über einen definierten Zeitraum, die Verwendbarkeitsfrist, zu gewährleisten. Die Bedeutung wird auch dadurch verdeutlicht, dass die Stabilitätsleitlinie Q1A „Stability Testing of New Drug Substances and Products“ das erste Thema war, welches im ...

Beitrag aus der Ausgabe 2/2026 der Zeitschrift pharmind
GDP-Inspektionen
Häufige Mängel bei der Qualifizierung von Logistikdienstleistern und wie man sie vermeidet – Fortsetzung*Vorheriger Teil s. Pharm. Ind. 87, Nr. 11, 1004–1008 (2025).
Die Aufgaben eines Großhändlers nach § 52a AMG sind in der Arzneimittelhandelsverordnung sowie in der EU-GDP-Leitlinie geregelt. Die zuständige Behörde überprüft die Einhaltung dieser Anforderungen im Rahmen von Inspektionen gemäß § 64 AMG.Im Zuge dieser GDP-Inspektionen stellt die Qualifizierung von Logistikdienstleistern einen wesentlichen Prüfpunkt dar. Dieser Aspekt gewinnt insb. dann an Bedeutung, wenn umfangreiche oder ...
Beitrag aus der Ausgabe 3/2026 der Zeitschrift pharmind
Pflanzliche Arzneimittel
Analytik und Qualitätssicherung – Teil 1
Dieser Beitrag gibt einen Überblick über die aktuellen Qualitätsanforderungen und analytischen Kontrollen an pflanzlichen (pfl.) Arzneimitteln in nationalen und europäischen Zulassungs- und Registrierungsverfahren. Nach § 4 Abs. 29 des deutschen Arzneimittelgesetzes (AMG) werden diese als Arzneimittel definiert, welche als Wirkstoff ausschließlich einen oder mehrere pflanzliche Stoffe oder eine oder mehrere pflanzliche ...
Top Themen

Beitrag aus der Ausgabe 7/2026 der Zeitschrift pharmind
Quo vadis Pharmaglas?
Die nächste Generation von Borosilikatglas für pharmazeutische Anwendungen
Die Entwicklung von Glas als Primärpackmittel für pharmazeutische Anwendungen nahm ihren Anfang mit dem Zusammentreffen zweier deutscher Pioniere in der Glasforschung. Der Chemiker Otto Schott (1851–1935) und der Physiker Ernst Abbe (1840–1905) begründeten 1879 eine Partnerschaft zur gemeinsamen Entwicklung von Hochleistungsgläsern [1]. 1911 folgte daraufhin die Entwicklung eines Borosilikatglases, Glas A, welches sich ...



Vorschau (Änderungen vorbehalten)
.jpg)
Beitrag aus der nächsten Ausgabe 8/2026 der Zeitschrift pharmind
(erscheint am 31.08.2026)
Gereinigtes Wasser für die Pharmalogistik | Eine Fallstudie bei einem Logistikdienstleister
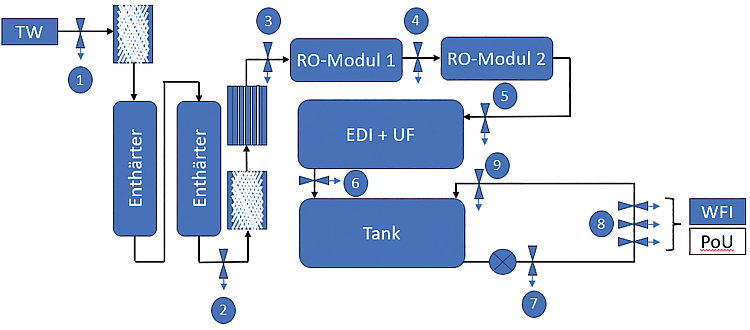
Die vorliegende Fallstudie analysiert die Planung, Errichtung und Qualifizierung einer Anlage zur Erzeugung, Lagerung und Verteilung von gereinigtem Wasser (Purified Water, PW). Im Mittelpunkt steht der konkrete Anwendungsfall eines Logistikdienstleisters in Ravensburg, der sich vom Good-Distribution-Practice(GDP)- zum Good-Manufacturing-Practice(GMP)-konformen Pharmadienstleister entwickelt. Am Standort werden Kryobehälter für biopharmazeutische Wirkstoffe für einen internationalen pharmazeutischen Hersteller gelagert, gereinigt und gewartet.