


Aseptische Herstellung steriler Arzneimittel
Faktoren zur Verhinderung einer Produktkontamination
Produktion
Key WordsAseptische Prozessierung | Kontaminationskontrolle | Sterilität | Barrier-Konzept | Automation
Zusammenfassung
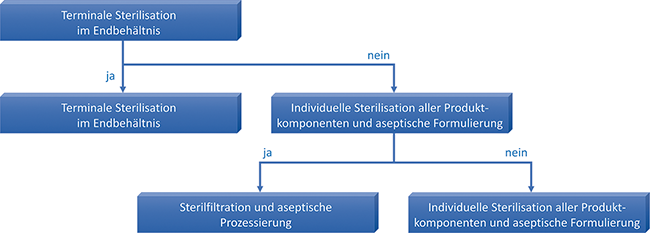
Wenn eine terminale Sterilisation nicht möglich ist, werden sterile Arzneimittel unter aseptischen Bedingungen hergestellt. Hierbei werden zum Schutz des Produkts vielfältige Maßnahmen zur Kontaminationskontrolle kombiniert. Den äußerst hohen Maßstäben an die Produktionsräume, -anlagen und -prozesse wird mit weitreichenden qualitätssichernden Instrumenten begegnet. Dabei ist es besonders wichtig, die Gefahr, die vom größten Kontaminationsfaktor – dem Personal – ausgeht, zu minimieren.
Korrespondenz:
Prof. Dr. Andreas Schmid, Hochschule Albstadt-Sigmaringen, Anton-Günther-Str. 51, 72488 Sigmaringen; E-Mail: schmida@hs-albsig.de
 | Prof. Dr. Andreas Schmid Andreas Schmid ist seit 2012 Professor an der Hochschule Albstadt-Sigmaringen. Seit 2013 ist er Dekan der Fakultät Life Sciences. Seine Lehrgebiete liegen in den Bereichen Steril- und Reinraumtechnik sowie Biotechnologie. Vor seinem Ruf an die Hochschule war er nach dem Studium der Biotechnologie und der Promotion am European Neuroscience Institute der Universität Göttingen mehrere Jahre als Leiter der |
Schließen Sie hier ein Abonnement ab und profitieren Sie von den vielseitigen Nutzungsmöglichkeiten.

