


Risikobasierte Festlegung von Monitoringpunkten im aseptischen Herstellbereich
Messen/Steuern/Regeln
Key WordsMonitoring | Aseptik | Isolator | RABS | risikobasiert
Zusammenfassung
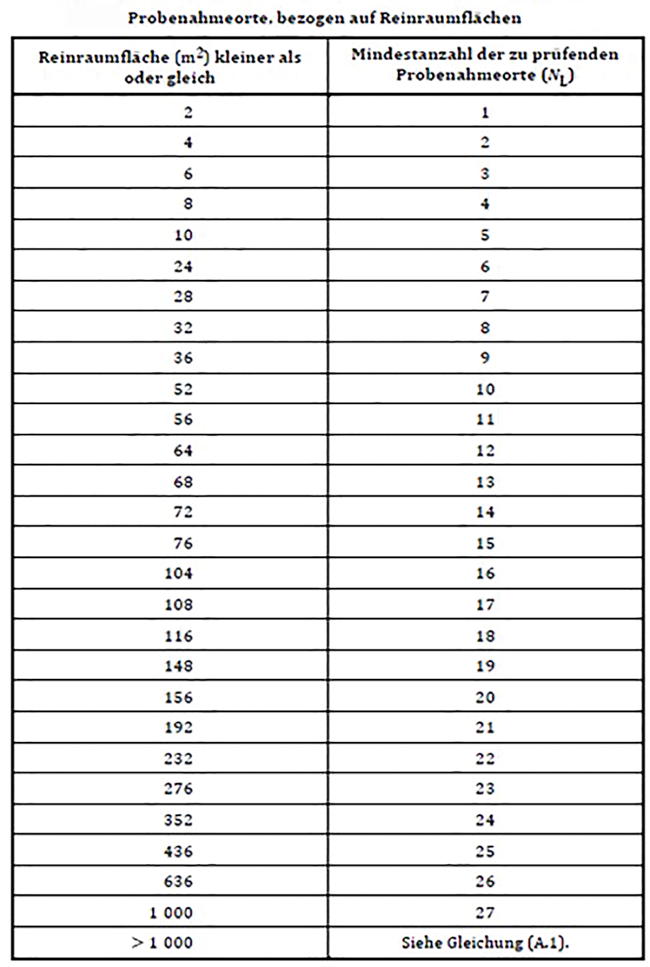
Die Sterilität ist das wichtigste Qualitätsmerkmal der Umgebung der Reinraumklasse A/ISO 5 zur Fertigung aseptischer Produkte. Die Hersteller von aseptischen Darreichungsformen müssen diese Qualität gewährleisten und die hierfür „kritischen Einflussfaktoren“ überwachen. Die Bewertung und Festlegung der Monitoringpunkte muss hierbei schon in einer frühen Projektphase erfolgen. Bei der risikobasierten Festlegung der Monitoringpunkte müssen unterschiedliche regulatorische Anforderungen berücksichtigt werden, die im Rahmen der Design-Phase des RABS- oder Isolator-Systems über die Design-FMEA und Mock-up bestätigt werden.
Korrespondenz:
Christian Gavranovic, PPT Pharma Process Technology GmbH, Neue Mainzer Str. 66–68, 60311 Frankfurt am Main; E-Mail: christian.gavranovic@pp-technology.de
 | Christian Gavranovic Christian Gavranovic ist studierter Maschinenbau-Ingenieur. Er startete seine berufliche Laufbahn bei der Boehringer Ingelheim GmbH als Process Engineer. Nach seinem Wechsel zur Biotest AG betreute er zuerst den Bereich der aseptischen Abfüllung und Gefriertrocknung als Quality Assurance Manager. Anschließend unterstützte er die Abteilung der Endkonfektionierung als Betriebsassistent. Ab |
Schließen Sie hier ein Abonnement ab und profitieren Sie von den vielseitigen Nutzungsmöglichkeiten.

