


Einführung in die sterile Herstellung
GMP-Praxis
Key Words Primärpackmittel | sterile Herstellung | aseptische Herstellung | sterile Produkte | Regularien
Zusammenfassung
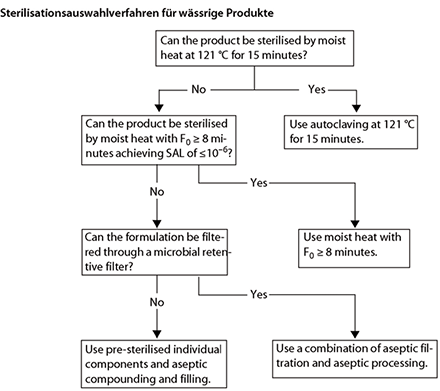
Sterile Produkte müssen frei von überlebensfähigen Keimen sein. Außerdem sind die Grenzwerte für Partikel und Endotoxine einzuhalten. Die Sterilität wird entweder durch Dampfsterilisation im Endbehältnis (Methode der Wahl), Sterilfiltration oder durch eine aseptische Herstellungsweise erreicht.
Der Beitrag gibt eine Einführung in die Grundlagen der Herstellung von sterilen Produkten. Die Unterschiede von steriler und aseptischer Herstellung werden erläutert und die gesetzlichen Grundlagen dargestellt.
Korrespondenz:
Dipl.-Ing. Ruven Brandes, Senator-Bauer-Straße 19, 30652 Hannover (Germany); e-mail: rbconsulting@gmx.eu
 | Ruven Brandes Ruven Brandes studierte Bioverfahrenstechnik an der FH Hannover und nahm 2001 seine Tätigkeit bei der Wirtschaftsgenossenschaft deutscher Tierärzte eG (WDT) auf. Er war maßgeblich an GMP-gerechten Umbauprojekten beteiligt. Seit 2006 ist Brandes Leiter Technik und Compliance Support technische QS bei der WDT. Zusätzlich umfasst sein Aufgabengebiet u. a. auch die Qualifizierung und die Reinigungsvalidierung in der pharmazeutischen Produktion. Er engagiert sich zudem in verschiedenen |
Schließen Sie hier ein Abonnement ab und profitieren Sie von den vielseitigen Nutzungsmöglichkeiten.

