


Fabrikplanung im GMP-Umfeld
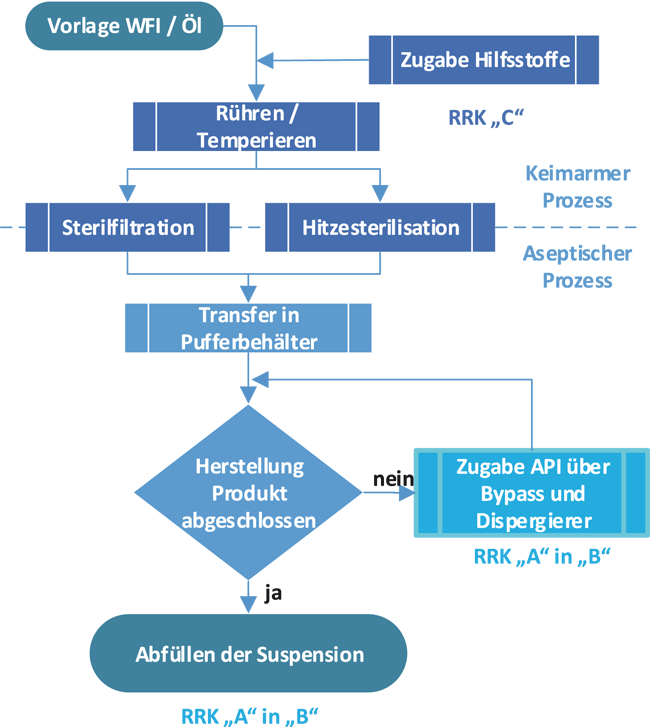
Beispiel einer aseptischen Ansatzbereitung
Key Words Fabrikplanung | Sterilherstellung | Aseptische Herstellung | HOAI | Sterile Suspension
Zusammenfassung
Fertigung und Abfüllung steriler Suspensionen für die Pharmaindustrie stellen verfahrenstechnische Herausforderungen dar. Bei Ansatzgrößen von 2 000 l müssen große Mengen pulverförmigen Wirkstoffs aseptisch gehandhabt, dispergiert sowie als Suspension zu einer Füllmaschine transferiert werden. Es gibt verschiedene Möglichkeiten, diese Prozesse technisch umzusetzen. Im Rahmen einer Fabrikplanung sind diese Optionen zu evaluieren, für die gewählte das Layout zu entwickeln und die Versorgersysteme auszulegen. Dieser Beitrag fokussiert sich auf Aspekte des Kernprozesses, die maßgeblich das Good-Manufacturing-Practice(GMP)-Layout sowie die technische Gebäudeausrüstung beeinflussen, und stellt exemplarisch den Einfluss einzelner Entscheidungen auf eine Fabrikplanung beim Tierarzneimittelhersteller aniMedica GmbH – a LIVISTO company dar.
Thomas Höltker · aniMedica GmbH – a LIVISTO company, Senden-Bösensell
Korrespondenz:
Christian Panhans, Head Office Nuremberg, Division Technology Facilities, M+W Central Europe GmbH – A Company of the M+W Group, Rollnerstr. 97, 90408 Nürnberg; e-mail: christian.panhans@mwgroup.net
Zusammenfassung
Fertigung und Abfüllung steriler Suspensionen für die Pharmaindustrie stellen verfahrenstechnische Herausforderungen dar. Bei Ansatzgrößen von 2 000 l müssen große Mengen pulverförmigen Wirkstoffs aseptisch gehandhabt, dispergiert sowie als Suspension zu einer
Schließen Sie hier ein Abonnement ab und profitieren Sie von den vielseitigen Nutzungsmöglichkeiten.

