


Handhabung von aseptisch hergestellten, hochaktiven Pulvern
Anforderungen in Bezug auf GMP und Arbeitsschutz
Key WordsAseptische Herstellung | Toxisch-aseptische Pulver | Vermeidung von Kreuzkontamination | Reinigung | Mitarbeiterschutz
Zusammenfassung
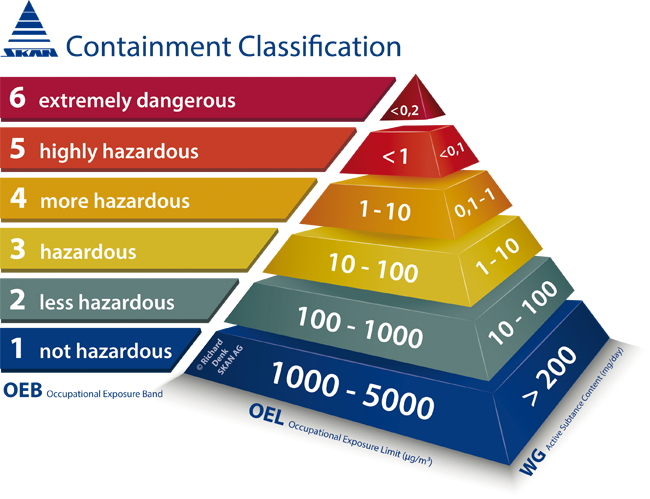
Die Integration des Produkttransfers von pulvrigen, sterilen toxischen Stoffen in einer Gesamtlösung von der Filtertrocknerentleerung bis hin zur sterilen Abfüllung des Wirkstoffes in Vials hat die SKAN AG zusammen mit Atec Pharmatechnik GmbH umgesetzt. In dem Gesamtprojekt wurden die Good-Manufacturing-Practice(GMP)-Anforderungen zur aseptischen und sicheren Handhabung des Wirkstoffes gemäß einem Quality Risk Management umgesetzt. So können die arbeitshygienischen Anforderungen zum Bedienerschutz zur Erreichung eines Occupational Exposure Levels (OEL) kleiner 1 μg/m3 eingehalten werden. Zur Vermeidung einer Kreuzkontamination bei der Herstellung von unterschiedlichen Wirkstoffen in einer Vielstoffanlage sind die Reinigungsanforderungen für aseptische Isolatoren gemäß dem Letter „Isolator Surfaces and Contamination Risk to Personnel and Patient“ der Parenteral Drug Association (PDA) und der darin enthaltenen Tabelle „Proposed EH&S and GMP surface limits for non-product contact surfaces and air limits of isolators“ in das Gesamtkonzept eingebunden.
Andreas Linz · Atec Pharmatechnik GmbH, Sörup
Korrespondenz
Richard Denk, Leiter Vertrieb Containment, SKAN AG, Binningerstrasse 116, 4123 Allschwil (Schweiz); e-mail: richard.denk@skan.ch
 | Richard Denk Richard Denk arbeitet bei der Firma SKAN AG mit Sitz in Allschwil (Schweiz) als Leiter Vertrieb Containment. Er gründete 2008 die Containment-Expertengruppe der ISPE D/A/CH, die im Nov. 2015 das Containment-Handbuch publiziert hat. Weiterhin ist er Autor beim Maas & Peither GMP-Verlag zum Thema Containment und Hygienic Design sowie einer der Autoren des ISPE Oral Solid Dosage Baseline Guide. Seit fast 20 Jahren |
Schließen Sie hier ein Abonnement ab und profitieren Sie von den vielseitigen Nutzungsmöglichkeiten.

