Ilmac Lausanne 2026: Inspiring the Future of Chemistry and Life Sciences
31.07.2026Ilmac ist eine exklusive internationale Fachveranstaltung, welche die gesamte Wertschöpfungskette der Chemie- und Life Sciences-Branche abbildet. Persönlich, inspirierend, wegweisend, exquisit. Der ...

Hitze: Der stille Killer im Krankenhaus
31.07.2026Die aktuelle Hitzewelle belastet alle Menschen – ganz besonders aber jene, die in einem Krankenhaus liegen oder dort arbeiten. Welche Folgen die Hitze für die Krankenhäuser hat und was dort ...
REALOGIS: Ruhrgebiet setzte sich im 1. Halbjahr 2026 an die Spitze der deutschen Logistik-Top-8
30.07.2026Die REALOGIS Unternehmensgruppe, Deutschlands führendes Beratungsunternehmen für Industrie- und Logistikimmobilien sowie Gewerbegrundstücke, registrierte im 1. Halbjahr 2026 an den von ihr ...

Blut spenden: „Es braucht nicht viel, um Gutes zu tun“
28.07.2026Es vergeht kein Tag in Deutschland, an dem nicht Blut und Blutplasma gebraucht wird – sei es für die Behandlung schwerer Krankheiten, nach Unfällen oder bei Operationen. Doch der Anteil der ...
Spatenstich für das Forschungsgebäude CARE-MED am Uniklinikum Erlangen
28.07.2026Einzigartige Fusion für KI-gestützte Präzisionsmedizin: Zwischen CESAR und CITABLE fügt sich auf dem Forschungscampus Nord schon bald CARE-MED ein, ein weiteres Forschungsgebäude des ...
Branchenpartner
meistgelesen

Beitrag aus der Ausgabe 1/2026 der Zeitschrift pharmind
Stabilitätsprüfung von Wirkstoffen und Arzneimitteln
Der Entwurf der neuen ICH-Leitlinie Q1
Stabilitätsuntersuchungen sind ein essenzieller Bestandteil der Arzneimittelentwicklung und -zulassung. Sie dienen dazu, die Qualität, Sicherheit und Wirksamkeit eines Arzneimittels über einen definierten Zeitraum, die Verwendbarkeitsfrist, zu gewährleisten. Die Bedeutung wird auch dadurch verdeutlicht, dass die Stabilitätsleitlinie Q1A „Stability Testing of New Drug Substances and Products“ das erste Thema war, welches im ...

Beitrag aus der Ausgabe 2/2026 der Zeitschrift Tech4Pharma
Standortbestimmung in der Pharmaproduktion
Fluoreszenztests zur Bewertung der Reinigbarkeit
Zur Beantwortung der Fragestellung „Wie gut lässt sich z. B. ein Isolator reinigen?“ haben sich sog. Fluoreszenztests bewährt. Dabei werden die relevanten Oberflächen mit einer unter UV-Beleuchtung stark fluoreszierenden Prüfsubstanz gezielt kontaminiert und anschließend einer exemplarischen Reinigung unterzogen. Danach werden die verbleibenden fluoreszierenden Restkontaminationen visuell meist im Bildvergleich zur initialen ...

Beitrag aus der Ausgabe 2/2026 der Zeitschrift cleanroom & processes
Schnee statt hochreinem Wasser
Medizin- und Pharmaprodukte sicher und ressourcenschonend reinigen
Das Spektrum an medizintechnischen Produkten und pharmazeutischen Erzeugnissen, die verpackt werden müssen, ist enorm vielfältig und stellt besondere Anforderungen an die Reinigung während der Herstellung. Dabei ist ein produktspezifisch definiertes Reinheitslevel zuverlässig zu erfüllen. Hinzu kommen regulatorische Vorgaben, die eingehalten werden müssen. Entsprechend ist ein Reinigungsprozess dann für die Herstellung von Medizintechnik- und ...
Top Themen

Beitrag aus der Ausgabe 7/2026 der Zeitschrift pharmind
Quo vadis Pharmaglas?
Die nächste Generation von Borosilikatglas für pharmazeutische Anwendungen
Die Entwicklung von Glas als Primärpackmittel für pharmazeutische Anwendungen nahm ihren Anfang mit dem Zusammentreffen zweier deutscher Pioniere in der Glasforschung. Der Chemiker Otto Schott (1851–1935) und der Physiker Ernst Abbe (1840–1905) begründeten 1879 eine Partnerschaft zur gemeinsamen Entwicklung von Hochleistungsgläsern [1]. 1911 folgte daraufhin die Entwicklung eines Borosilikatglases, Glas A, welches sich ...




Beitrag aus der Ausgabe 2/2026 der Zeitschrift Tech4Pharma
Einstieg in Continuous Manufacturing
Technologische, prozessuale und entwicklungsrelevante Aspekte der kontinuierlichen Feuchtgranulation
In der Pharmaindustrie werden Granulationsprozesse zunehmend unter den Aspekten der Effizienzsteigerung und Kostenoptimierung betrachtet. Ziel der Feuchtgranulation ist es, aus feinem Pulver gröbere Agglomerate zu erzeugen. Agglomerate können aus festen und trockenen Körnern bestehen, wobei jedes Korn ein Agglomerat aus Pulverpartikeln mit genügend Festigkeit darstellt. Granulate können direkt als Arzneimittel verwendet werden oder dienen als ...



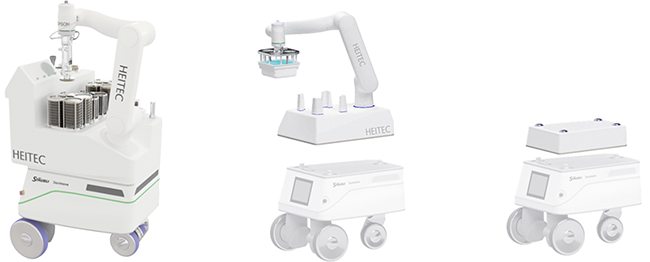
Beitrag aus der Ausgabe 2/2026 der Zeitschrift cleanroom & processes
Innovationen im pharmazeutischen Reinraum
Kontaktlose Authentifizierung, mobile Robotik und intelligente Bedienkonzepte für die Pharmaproduktion
In hochreinen Produktionsbereichen der Pharmaindustrie steht die Vermeidung von Kontaminationen an oberster Stelle. Das größte Risiko geht dabei vom Menschen aus, weshalb Reinräume nur von autorisiertem Fachpersonal betreten werden dürfen. Gleichzeitig erfordern moderne Herstellprozesse eine lückenlose Kontrolle und Dokumentation, um Qualität und Compliance – etwa mit den GMP-Regularien – sicherzustellen. Traditionelle Verfahren ...


Vorschau (Änderungen vorbehalten)
.jpg)
Beitrag aus der nächsten Ausgabe 8/2026 der Zeitschrift pharmind
(erscheint am 31.08.2026)
Gereinigtes Wasser für die Pharmalogistik | Eine Fallstudie bei einem Logistikdienstleister
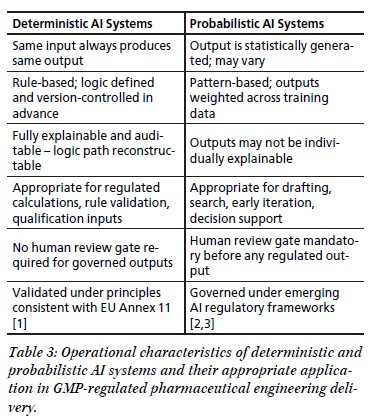
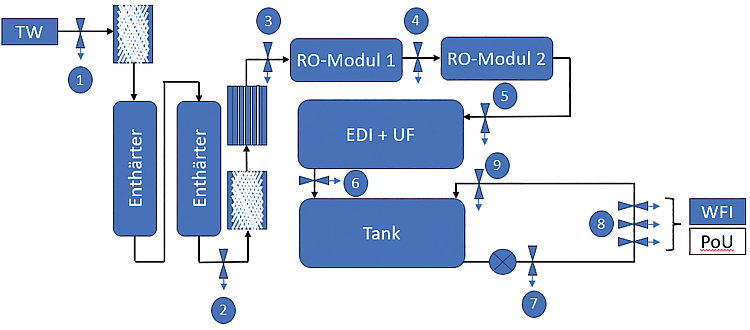
Die vorliegende Fallstudie analysiert die Planung, Errichtung und Qualifizierung einer Anlage zur Erzeugung, Lagerung und Verteilung von gereinigtem Wasser (Purified Water, PW). Im Mittelpunkt steht der konkrete Anwendungsfall eines Logistikdienstleisters in Ravensburg, der sich vom Good-Distribution-Practice(GDP)- zum Good-Manufacturing-Practice(GMP)-konformen Pharmadienstleister entwickelt. Am Standort werden Kryobehälter für biopharmazeutische Wirkstoffe für einen internationalen pharmazeutischen Hersteller gelagert, gereinigt und gewartet.
Beitrag aus der nächsten Ausgabe 3/2026 der Zeitschrift Tech4Pharma
(erscheint am 09.09.2026)
Fülltechnologie für flüssige, sterile Formen / Rüsten – Betreiben – Füllsystemtypen und deren Eigenschaften – Teil 2
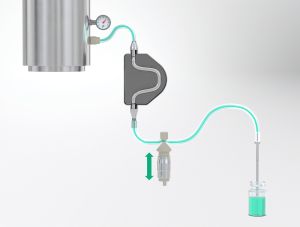
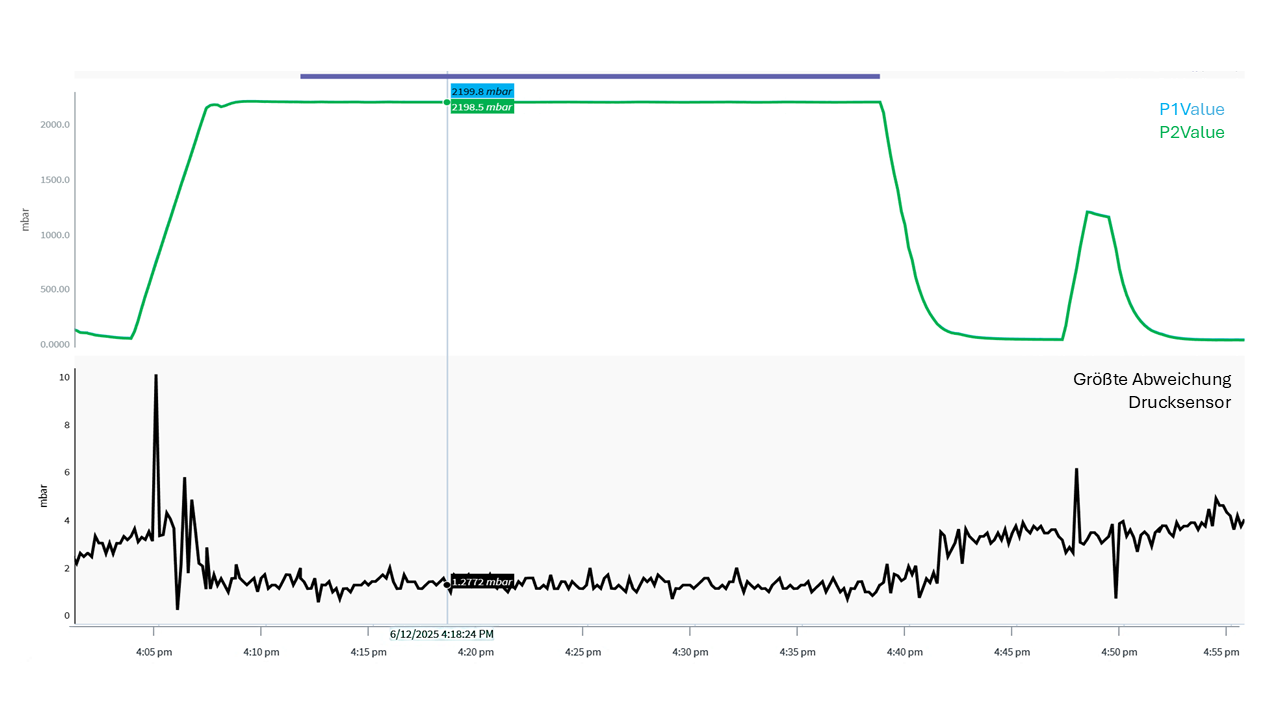
Im ersten Teil dieses Fachbeitrags wurden die regulatorischen Vorgaben für das Abfüllen flüssiger Pharmazeutika sowie das aseptische Rüsten gemäß des EU-GMP Annex 1 erläutert. Weiterhin wurden die Grundlagen des Füllprozesses und das Strömungsverhalten von Fluiden beschrieben. Dieser zweite Teil behandelt den Aufbau und die Funktionsweise gängiger Fülltechnologien. Zusätzlich werden Möglichkeiten zur Reduktion von Produktverlust vor, während und nach der Produktion aufgezeigt. Abschließend werden Anforderungen verschiedener Produktklassen von Medikamenten erläutert sowie die damit einhergehende Wahl eines geeigneten Füllsystems begründet.
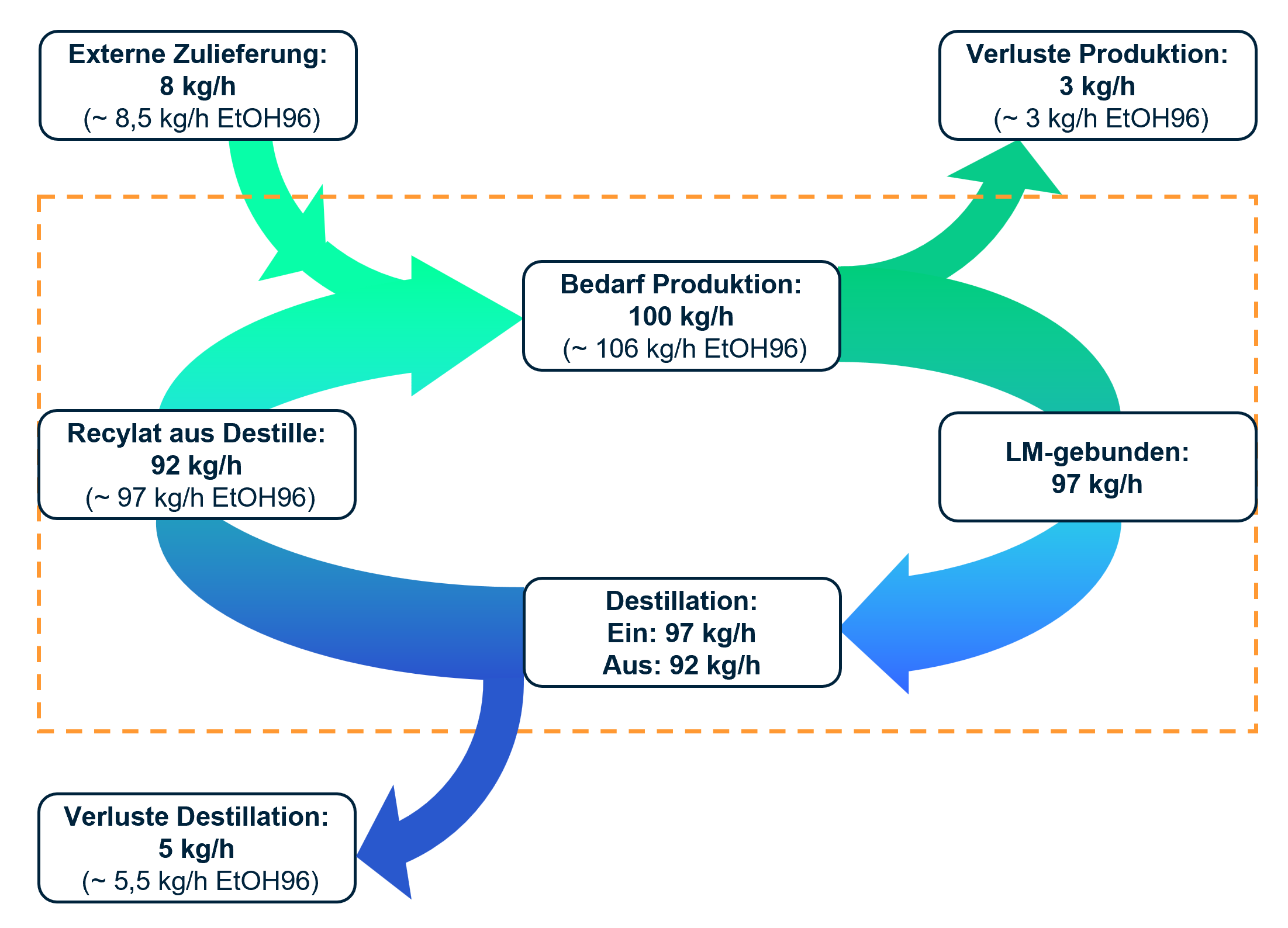
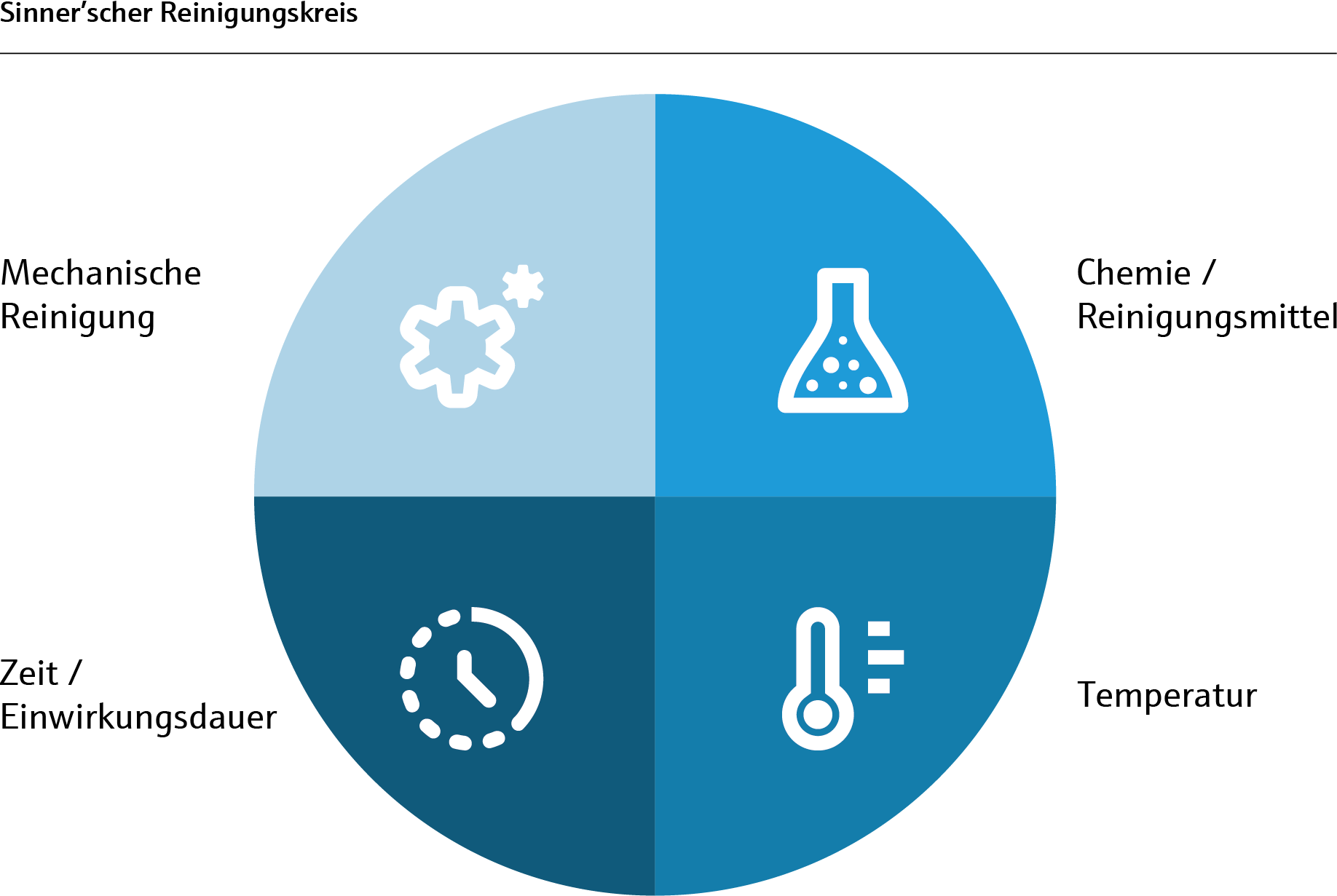

Beitrag aus der nächsten Ausgabe 03/2026 der Zeitschrift cleanroom & processes
(erscheint am 18.09.2026)
Trockenraum und Mensch - Wechselwirkungen und Beeinflussung
Die Produktion von Batteriezellen erfordert Rein- und Trockenräume mit hoher Partikelreinheit und extrem niedriger Luftfeuchte. Während die Technik solcher Umgebungen gut beschrieben ist, ist der Einfluss des Personals auf den Taupunkt sowie die Rückwirkung des trockenen Klimas auf die Mitarbeitenden kaum untersucht. Im Rein- und Trockenraumlabor des Fraunhofer IPA wurden Versuche zur durch Personen verursachten Veränderung des Taupunkts durchgeführt. Auf Basis der Ergebnisse wurden Verhaltensregeln sowie organisatorische und technische Maßnahmen abgeleitet, die sowohl die Stabilität des Trockenraumklimas als auch den Gesundheitsschutz unterstützen.