Adipositas: Eine verkannte und unterschätzte Erkrankung
23.10.2025Dabei gilt sie längst als eine der größten gesundheitlichen und gesundheitspolitischen Herausforderungen weltweit – die Zahl der Betroffenen auch in Deutschland steigt. Auf dem Europäischen ...

Wie Innovation Krankheit besiegt: Leukämien
20.10.2025Früher bestand die Behandlung von Leukämie-Erkrankungen in erster Linie aus Chemo- und Stammzelltherapie. Heute gibt es „mehr Optionen, mehr Personalisierung, mehr Chancen auf Heilung“, ...
Globale Registerdaten zeigen signifikanten Gesamtüberlebensvorteil bei Patienten mit fortgeschrittenem kutanem T-Zell-Lymphom (CTCL), die mit POTELIGEO (Mogamulizumab) behandelt wurden
23.10.2025Kyowa Kirin International (KKI), eine hundertprozentige Tochtergesellschaft des japanischen Speciality-Pharmaunternehmens Kyowa Kirin Co., Ltd., und der University Hospitals Birmingham NHS Foundation ...
Wilmington PharmaTech setzt Expansion mit bedeutender Investition von Curewell Capital fort
23.10.2025Wilmington PharmaTech, ein in den USA ansässiges Auftragsforschungs-, Entwicklungs- und Produktionsunternehmen (CRDMO), das sich auf die kundenspezifische Entwicklung und Herstellung von ...
Branchenpartner
meistgelesen

Beitrag aus der Ausgabe 5/2025 der Zeitschrift pharmind
Stabilitätsstudien
Planung, Durchführung und Dokumentation – Teil 1
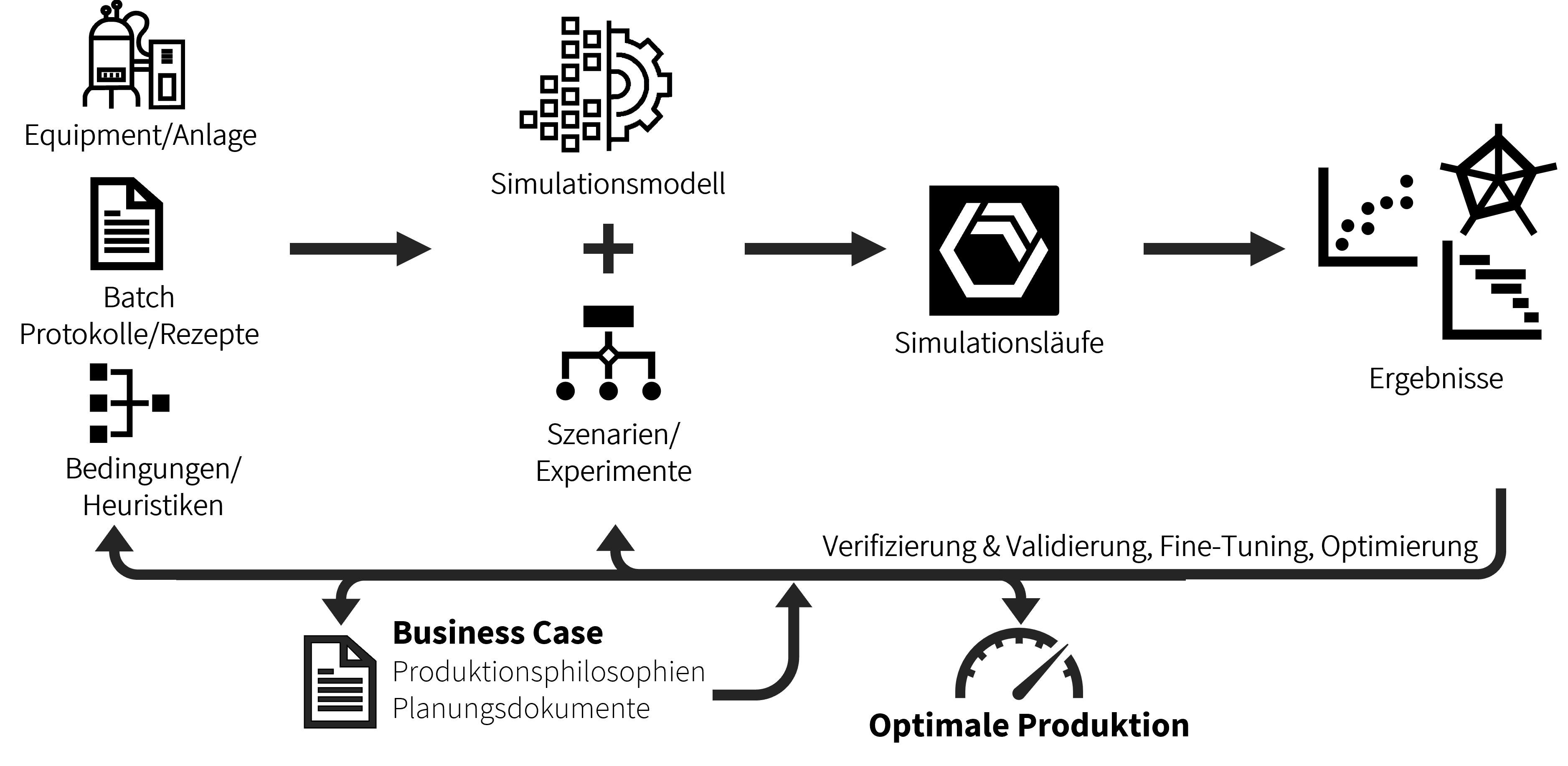
Der allgemeine Ablauf einer Stabilitätsstudie in der Zulassungs- und Vermarktungsphase ist in Abb. 1 schematisch dargestellt. Nachfolgend wird kurz auf die einzelnen Schritte eingegangen. Die genaue Durchführung ist detailliert in entsprechenden Standard Operating Procedures (SOPs) oder Arbeitsanweisungen zu beschreiben.Vor Beginn der Stabilitätsstudie wird das Studiendesign in einem Stabilitätsprotokoll (Stability Protocol) festgehalten. ...

Beitrag aus der Ausgabe 2/2025 der Zeitschrift Tech4Pharma
Temperaturverteilungsmessung
Herausforderungen bei einem Tieftemperatur-Mapping
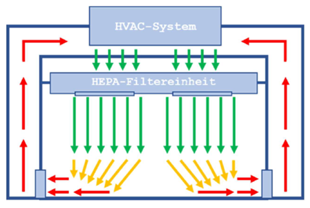
Die Temperatur- und Klimaverteilungsmessung, kurz Mapping genannt, bildet das zentrale Element einer Lager- oder Transportqualifizierung. Im EU-Leitfaden zur Good Distribution Practice (GDP) [1] wird die Durchführung einer Verteilungsstudie vor Inbetriebnahme und bei wesentlichen Veränderungen unter repräsentativen Bedingungen gefordert. Die Basis dafür bildet eine entsprechende Risikobetrachtung. Näheres dazu kann in [2] nachgelesen ...
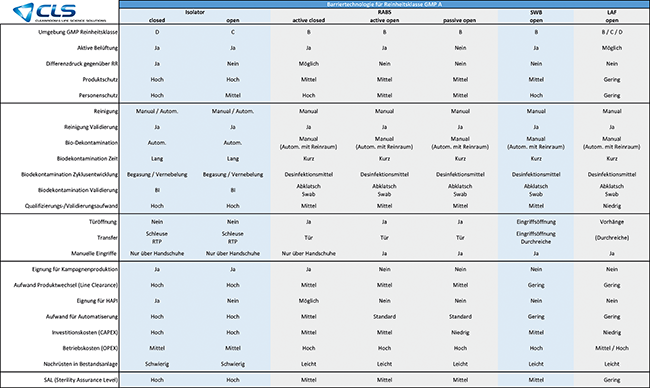
Beitrag aus der Ausgabe 2/2025 der Zeitschrift cleanroom & processes
Barrieretechnologie im GMP-Bereich
Worauf es ankommt – Teil 2*Teil 1 dieses Beitrags s. cleanroom & processes 2025; 4(1): 12-17.
Jede Barrieretechnologie muss den regulatorischen Vorschriften entsprechen, die durch die verschiedenen Aufsichtsbehörden festgelegt sind. Dies umfasst sowohl die GMP-Anforderungen (z. B. Produktsicherheit, Verhinderung von Kreuzkontamination, Sterilität, Reinigbarkeit, Validierung) als auch die Einhaltung von Umwelt- und Sicherheitsvorschriften (z. B. Schutz des Bedienpersonals).Die Reinigbarkeit von Barrieresystemen ist ein ...
Top Themen

Beitrag aus der Ausgabe 9/2025 der Zeitschrift pharmind
Optimizing Cleaning Validation Practices
In Complex Contract Development Manufacturing Environments
The scope of cleaning validation (CV), as outlined by global regulatory authorities, encompasses the documented evidence that cleaning procedures consistently and effectively remove chemical, microbiological, and particulate contaminants to predefined and acceptable levels. Regulatory bodies such as the FDA and EMA emphasize that cleaning validation must demonstrate the removal of residues from active pharmaceutical ingredients (APIs), ...




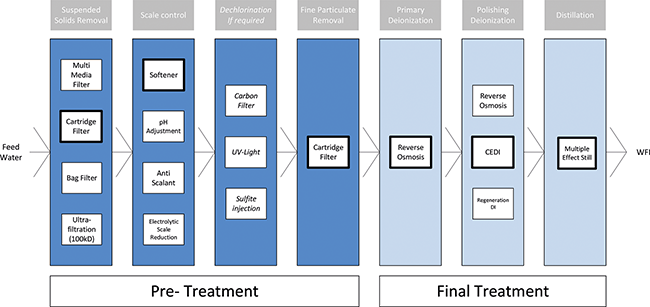
Beitrag aus der Ausgabe 3/2025 der Zeitschrift Tech4Pharma
Serie: Energetische Analyse von WFI-Systemen
Teil 1: WFI-Erzeugeranlagen – Funktionen aus energetischer Sicht
Im zweiten Serienteil werden standortbezogene Faktoren sowie Kriterien des Detail-Engineerings der Anlagen betrachtet, die für die Entscheidungsfindung von Bedeutung sind. Diese Faktoren können einen wesentlichen oder nur geringen Einfluss auf die Energieeffizienz der Herstellung von Wasser für Injektionszwecke (WFI) haben. Des Weiteren erfolgt eine Bewertung der genannten Faktoren hinsichtlich ihrer Auswirkungen auf die Energieeffizienz. ...
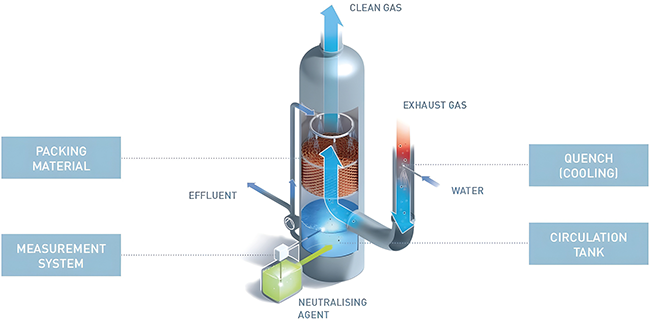
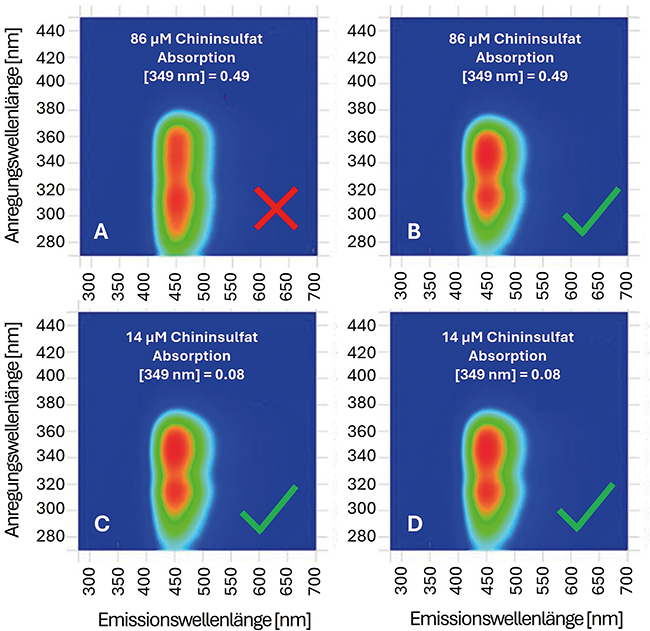
Beitrag aus der Ausgabe 3/2025 der Zeitschrift cleanroom & processes
Filtergängige Bakterien
Risiko bei der aseptischen Herstellung
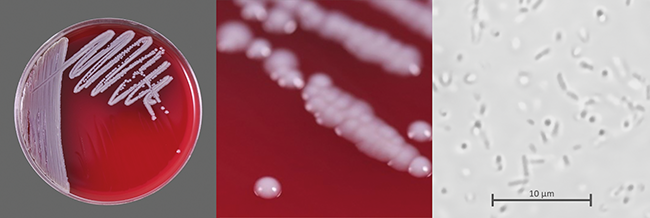
Die Sterilfiltration ist ein kritischer Prozess in der aseptischen Herstellung steriler Produkte, dessen Ziel die Entfernung von Mikroorganismen ist. Eine verbreitete Annahme ist, dass ein intakter Filter grundsätzlich alle Mikroorganismen zurückhält. Es gibt aber sehr wohl Mikroorganismen, die unter gewissen Umständen auch einen intakten und Pre-Use-Post-Sterilisation-Integrity-Testing(PUPSIT)-geprüften Filter passieren können. Biologische ...
Vorschau (Änderungen vorbehalten)
Beitrag aus der nächsten Ausgabe 11/2025 der Zeitschrift pharmind
(erscheint am 28.11.2025)
Arzneimittelengpässe im Fokus | Rechtslage, Reformbedarf und praktische Herausforderungen für die Industrie
Mit ungewöhnlicher Deutlichkeit attestiert der Europäische Rechnungshof der Europäischen Union in seinem Sonderbericht Nr. 19/2025 ein strukturelles Versagen bei der Bewältigung von Arzneimittelengpässen: Die Union verfügt trotz jahrelanger Bemühungen über kein wirksames System zur Vermeidung und Bewältigung von Arzneimittelengpässen. Dieser Fachbeitrag ordnet diese Kritik rechtlich und thematisch ein. Er zeichnet die Entwicklungen des unionsrechtlichen Rahmens seit 2011 nach, analysiert die strukturellen Defizite de lege lata und prüft, inwieweit die geplanten Reformen des „Pharmaceutical Package“ die vom Rechnungshof aufgezeigten Missstände tatsächlich beheben können. Für die pharmazeutische Industrie ergeben sich daraus erhebliche Konsequenzen: verschärfte Meldepflichten, neue Anforderungen an Lieferketten- Compliance und die Notwendigkeit, Versorgungssicherheit als festen Bestandteil regulatorischer Strategie zu begreifen. Versorgungssicherheit wird zu Rechtspflicht – und zum zentralen Kriterium künftiger Regulierung und Marktaufsicht.



Beitrag aus der nächsten Ausgabe 4/2025 der Zeitschrift Tech4Pharma
(erscheint am 14.11.2025)
Innovative Druckbildinspektions-Systeme / Sicherheit, Effizienz und Nachhaltigkeit für die Produktion von pharmazeutischen Packmitteln
Verpackungen von pharmazeutischen Erzeugnissen müssen höchste Anforderungen an Qualität, Sicherheit und Lesbarkeit erfüllen. Mithilfe eines passenden Druckinspektionssystems kann durch automatisierte und objektive Prüfungen entlang des kompletten Produktionsprozesses sichergestellt werden, dass alle gedruckten Informationen frei von Fehlern, vollständig und korrekt sind. Möglichkeiten zur Prüfung gibt es vom Artwork-Check über die Inspektion beim Druck bis hin zur Wareneingangskontrolle. Die Ergebnisse sind jederzeit reproduzierbar und dokumentiert – wichtig in regulierten Branchen und im Auditfall. Größter Innovationstreiber ist hier eine vortrainierte Künstliche Intelligenz, die die Effizienz und Benutzerfreundlichkeit des Prüfprozesses deutlich steigert. Die Systeme tragen aber auch durch die frühe Entdeckung von Fehlern zu erheblichen Ressourceneinsparungen und einer effizienten, schlanken Produktion bei.
Beitrag aus der nächsten Ausgabe 04/2025 der Zeitschrift cleanroom & processes
(erscheint am 07.11.2025)
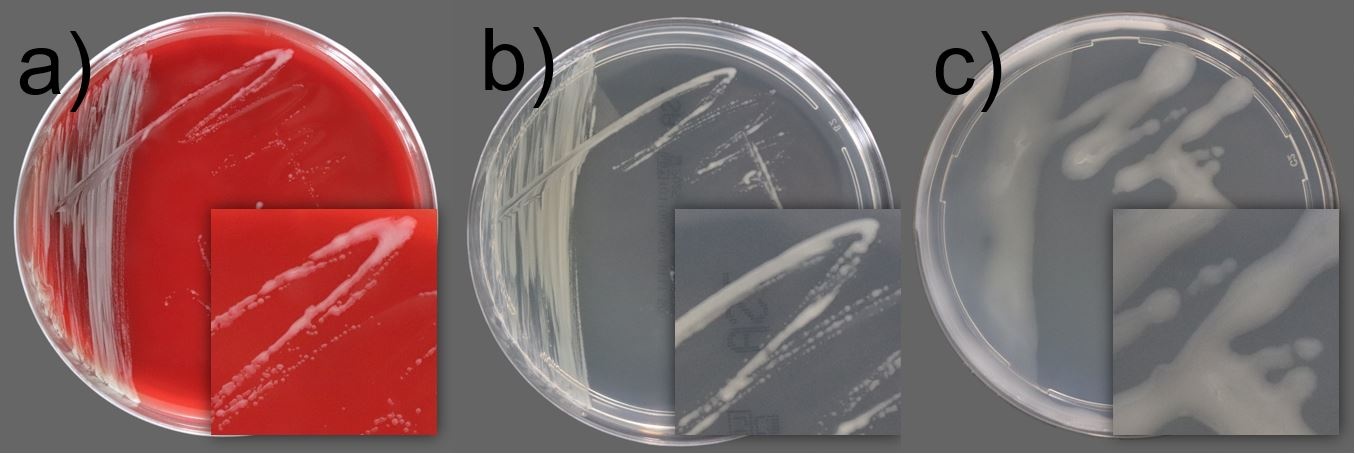
Is EN 16615 suitable for cleanrooms?
Disinfectants that are used in an environment where good manufacturing practice (GMP) is employed are required to be validated to demonstrate that they can reduce typical and anticipated bioburden to an acceptable level. EN 16615 provides the test method closest to the practical use of disinfectants because it incorporates the action of wiping the disinfectant onto a surface. As the method was initially intended to test disinfectant and antiseptic wipes in the medical area, this study has been conducted using EN 16615 methodology with modifications to include cleanroom-relevant surfaces (stainless steel and vinyl flooring) and organisms; Staphylococcus epidermidis (bacteria) and Bacillus subtilis (spore former). Testing has been performed to represent small and large surface disinfection with wipes and mops and a variety of chemistries with varying modes of biocidal action. The study results demonstrated that reductions of >3 log bacteria and >2 log spores were achieved, in compliance to USP <1072> Disinfectants and Antiseptics. These findings support the suitability of EN 16615, with relevant adaptations, for disinfectant validation in GMP-compliant cleanroom environments.