Doppeltes Leid: Frauen mit seltenen Erkrankungen
26.09.2025Frauen mit seltenen Erkrankungen leiden doppelt. Zum einen ist der Alltag mit einer solchen Krankheit oft geprägt von langen diagnostischen Irrwegen, unzureichender Therapieoptionen und einem ...

Chemie & Pharma Summit 2025
26.09.2025Kriege, Handelskonflikte, Unsicherheit: Die Weltwirtschaft schwächelt – und Deutschland droht das dritte Rezessionsjahr in Folge. „Wir sind nominell noch die drittgrößte Volkswirtschaft, ...
MOLDAWIEN BUSINESS WEEK 2025
25.09.2025Die Moldova Business Week (MBW) 2025 endete mit einer starken Botschaft an globale Investoren: Moldawien ist offen für Geschäfte und bereit zu wachsen. Die Jubiläumsausgabe des wichtigsten ...
EVACO wird DATEV-Partner – smarte Finanzanalysen jetzt im DATEV-Marktplatz
26.09.2025EVACO ist mit der Lösung EVACO DATEV Connect offizieller DATEV-Marktplatz Schnittstellen Partner. Damit verbinden sich jahrzehntelange DATEV-Expertise mit modernster Business-Analytics-Kompetenz.
Branchenpartner
meistgelesen

Beitrag aus der Ausgabe 5/2025 der Zeitschrift pharmind
Stabilitätsstudien
Planung, Durchführung und Dokumentation – Teil 1
Der allgemeine Ablauf einer Stabilitätsstudie in der Zulassungs- und Vermarktungsphase ist in Abb. 1 schematisch dargestellt. Nachfolgend wird kurz auf die einzelnen Schritte eingegangen. Die genaue Durchführung ist detailliert in entsprechenden Standard Operating Procedures (SOPs) oder Arbeitsanweisungen zu beschreiben.Vor Beginn der Stabilitätsstudie wird das Studiendesign in einem Stabilitätsprotokoll (Stability Protocol) festgehalten. ...

Beitrag aus der Ausgabe 1/2025 der Zeitschrift Tech4Pharma
Wer hat Angst vor Annex 1?
Herausforderungen und Lösungen bei der Produktionsanpassung an neue GMP-Richtlinien
Auch wenn sich die Generalüberarbeitung von Annex 1 [1] lange ankündigte, stehen Hersteller noch immer vor vielen ungeklärten Fragen zur Umsetzung. Sie müssen sowohl Bauteile als auch das Handling und alle dafür benötigten Werkzeuge und Mittel überprüfen. Die Zeit drängt, weil unter der Maßgabe der neuen Richtlinien bereits Audits stattfinden. Als Konsequenz können Auditoren aufwendige Umrüstungen verordnen, die sie sogar als Bedingung ...
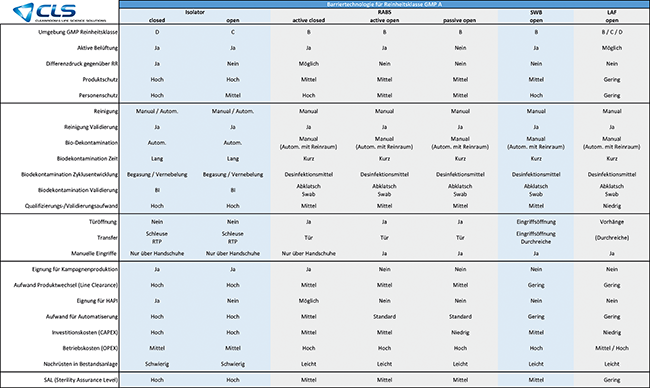
Beitrag aus der Ausgabe 2/2025 der Zeitschrift cleanroom & processes
Barrieretechnologie im GMP-Bereich
Worauf es ankommt – Teil 2*Teil 1 dieses Beitrags s. cleanroom & processes 2025; 4(1): 12-17.
Jede Barrieretechnologie muss den regulatorischen Vorschriften entsprechen, die durch die verschiedenen Aufsichtsbehörden festgelegt sind. Dies umfasst sowohl die GMP-Anforderungen (z. B. Produktsicherheit, Verhinderung von Kreuzkontamination, Sterilität, Reinigbarkeit, Validierung) als auch die Einhaltung von Umwelt- und Sicherheitsvorschriften (z. B. Schutz des Bedienpersonals).Die Reinigbarkeit von Barrieresystemen ist ein ...
Top Themen

Beitrag aus der Ausgabe 8/2025 der Zeitschrift pharmind
CO2-Reduktion im Mittelstand
Nachhaltigkeit praktisch und erfolgreich umgesetzt am Beispiel von PEKANA
Mit der UN-Klimakonferenz 2015 in Paris und dem daraus hervorgegangenen globalen Klimaabkommen ist die Bedeutung der unternehmerischen Nachhaltigkeit weiter gestiegen. Der europäische Green Deal gibt das Ziel vor: eine moderne, ressourceneffiziente und wettbewerbsfähige Wirtschaft, die bis 2050 keine Netto-Treibhausgase mehr ausstößt und ihr Wachstum von Ressourcennutzung abkoppelt [1]. Berichtspflichten u. a. aus der Europäischen ...

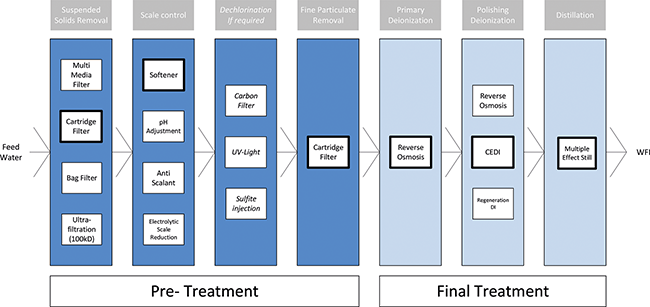
Beitrag aus der Ausgabe 3/2025 der Zeitschrift Tech4Pharma
Serie: Energetische Analyse von WFI-Systemen
Teil 1: WFI-Erzeugeranlagen – Funktionen aus energetischer Sicht
Im zweiten Serienteil werden standortbezogene Faktoren sowie Kriterien des Detail-Engineerings der Anlagen betrachtet, die für die Entscheidungsfindung von Bedeutung sind. Diese Faktoren können einen wesentlichen oder nur geringen Einfluss auf die Energieeffizienz der Herstellung von Wasser für Injektionszwecke (WFI) haben. Des Weiteren erfolgt eine Bewertung der genannten Faktoren hinsichtlich ihrer Auswirkungen auf die Energieeffizienz. ...
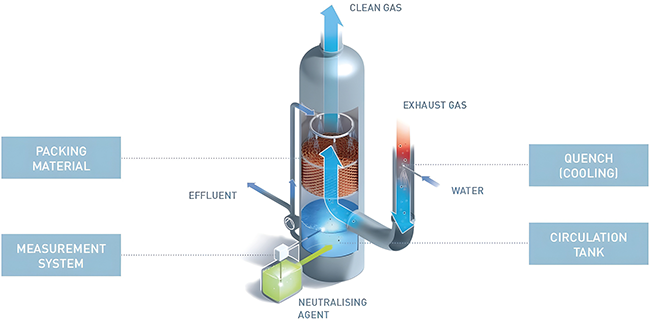
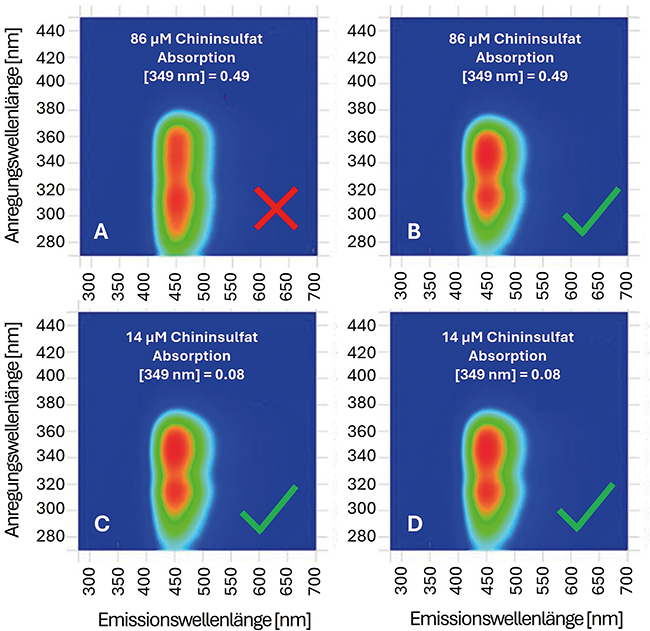
Beitrag aus der Ausgabe 3/2025 der Zeitschrift cleanroom & processes
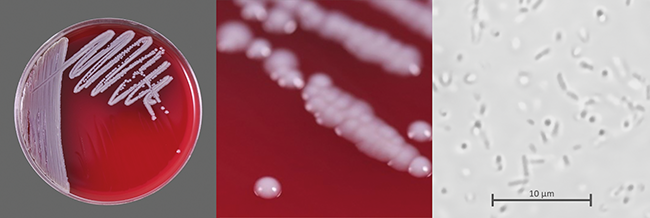
Filtergängige Bakterien
Risiko bei der aseptischen Herstellung
Die Sterilfiltration ist ein kritischer Prozess in der aseptischen Herstellung steriler Produkte, dessen Ziel die Entfernung von Mikroorganismen ist. Eine verbreitete Annahme ist, dass ein intakter Filter grundsätzlich alle Mikroorganismen zurückhält. Es gibt aber sehr wohl Mikroorganismen, die unter gewissen Umständen auch einen intakten und Pre-Use-Post-Sterilisation-Integrity-Testing(PUPSIT)-geprüften Filter passieren können. Biologische ...
Vorschau (Änderungen vorbehalten)
Beitrag aus der nächsten Ausgabe 10/2025 der Zeitschrift pharmind
(erscheint am 31.10.2025)
Pflanzliche Arzneimittel unter Druck | Markt, Regulierung und Zukunftsperspektiven – Teil 1
Pflanzliche Arzneimittel sind ein wesentlicher Bestandteil der Selbstmedikation und fest im deutschen Gesundheitsmarkt verankert. Trotz hoher Beliebtheit und langer Tradition geraten sie zunehmend unter Druck: Der Markt stagniert, Zulassungszahlen sinken und regulatorische Hürden steigen. Besonders betroffen sind kleine und mittelständische Hersteller, die durch hohe Kosten und zunehmenden bürokratischen Aufwand in ihrer Wettbewerbsfähigkeit eingeschränkt werden. Neben ökonomischen Faktoren gefährden regulatorische Änderungen – wie der geplante Wegfall des Well-Established Use, die Überarbeitung der Variation Regulation, die Neubewertung von Ethanol oder die Umsetzung der EU-Kommunalabwasserrichtlinie – die Zukunftsfähigkeit dieser Arzneimittel. Lösungsansätze erfordern eine differenzierte Regulierung, die ihre Bedeutung im Gesundheitswesen langfristig sichert.


Beitrag aus der nächsten Ausgabe 4/2025 der Zeitschrift Tech4Pharma
(erscheint am 14.11.2025)
Innovative Druckbildinspektions-Systeme / Sicherheit, Effizienz und Nachhaltigkeit für die Produktion von pharmazeutischen Packmitteln
Verpackungen von pharmazeutischen Erzeugnissen müssen höchste Anforderungen an Qualität, Sicherheit und Lesbarkeit erfüllen. Mithilfe eines passenden Druckinspektionssystems kann durch automatisierte und objektive Prüfungen entlang des kompletten Produktionsprozesses sichergestellt werden, dass alle gedruckten Informationen frei von Fehlern, vollständig und korrekt sind. Möglichkeiten zur Prüfung gibt es vom Artwork-Check über die Inspektion beim Druck bis hin zur Wareneingangskontrolle. Die Ergebnisse sind jederzeit reproduzierbar und dokumentiert – wichtig in regulierten Branchen und im Auditfall. Größter Innovationstreiber ist hier eine vortrainierte Künstliche Intelligenz, die die Effizienz und Benutzerfreundlichkeit des Prüfprozesses deutlich steigert. Die Systeme tragen aber auch durch die frühe Entdeckung von Fehlern zu erheblichen Ressourceneinsparungen und einer effizienten, schlanken Produktion bei.
Beitrag aus der nächsten Ausgabe 04/2025 der Zeitschrift cleanroom & processes
(erscheint am 07.11.2025)
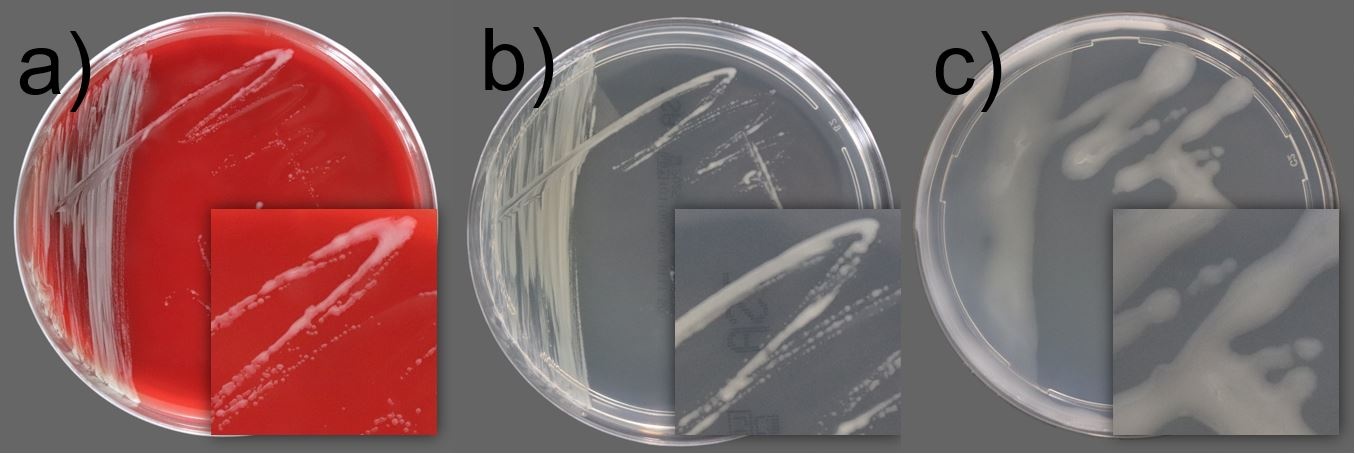
Is EN 16615 suitable for cleanrooms?
Disinfectants that are used in an environment where good manufacturing practice (GMP) is employed are required to be validated to demonstrate that they can reduce typical and anticipated bioburden to an acceptable level. EN 16615 provides the test method closest to the practical use of disinfectants because it incorporates the action of wiping the disinfectant onto a surface. As the method was initially intended to test disinfectant and antiseptic wipes in the medical area, this study has been conducted using EN 16615 methodology with modifications to include cleanroom-relevant surfaces (stainless steel and vinyl flooring) and organisms; Staphylococcus epidermidis (bacteria) and Bacillus subtilis (spore former). Testing has been performed to represent small and large surface disinfection with wipes and mops and a variety of chemistries with varying modes of biocidal action. The study results demonstrated that reductions of >3 log bacteria and >2 log spores were achieved, in compliance to USP <1072> Disinfectants and Antiseptics. These findings support the suitability of EN 16615, with relevant adaptations, for disinfectant validation in GMP-compliant cleanroom environments.